
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold nanoparticles with tailored size through ligand modification for catalytic applications†
Nidhi
Kapil
 a,
Fabio
Cardinale
a,
Tobias
Weissenberger
a,
Panagiotis
Trogadas
a,
T. Alexander
Nijhuis
b,
Michael M.
Nigra
*c and
Marc-Olivier
Coppens
*a
a,
Fabio
Cardinale
a,
Tobias
Weissenberger
a,
Panagiotis
Trogadas
a,
T. Alexander
Nijhuis
b,
Michael M.
Nigra
*c and
Marc-Olivier
Coppens
*a
aCentre for Nature Inspired Engineering and Department of Chemical Engineering, University College London, London WC1E 7JE, UK. E-mail: m.coppens@ucl.ac.uk
bSABIC Europe, Geleen, Limburg 6167 RD, The Netherlands
cDepartment of Chemical Engineering, University of Utah, Salt Lake City UT 84112, USA. E-mail: michael.nigra@utah.edu
First published on 24th September 2021
Abstract
The active sites of catalysts can be tuned by using appropriate organic moieties. Here, we describe a facile approach to synthesise gold nanoparticles (AuNPs) using various Au(I) precursors. The core size of these AuNPs can be precisely tailored by varying the steric hindrance imposed by bound ligands. An interesting relationship is deduced that correlates the steric hindrance around the metal to the final size of the nanoparticles. The synthesised AuNPs are immobilised onto TS-1 zeolite (Au/TS-1) with minimal change in the final size of the AuNPs. The catalytic performance of Au/TS-1 catalyst is evaluated for the direct gas phase epoxidation of propylene with hydrogen and oxygen, an environmentally friendly route to produce propylene oxide. The results indicate that smaller AuNPs exhibit enhanced catalytic activity and selectivity. Furthermore, this synthetic approach is beneficial when tailored synthesis of gold nanoparticles of specific sizes is required.
Gold nanoparticles (AuNPs) have been deployed in a variety of applications, including catalysis, medicine, electronics, sensing and labelling.1–4 The physical and chemical properties of these nanoparticles can be tailored by changing the particle size, shape, dispersion, and surface properties.5–7 Small AuNPs (<10 nm) increase both the available surface area and the fraction of gold atoms with low coordination number, often resulting in enhanced catalytic activity.8–10 Since the discoveries of Hutchings and Haruta that AuNPs are catalytically active for several important reactions, their synthesis has attracted even broader interest.2–4,11–13 The synthesis of these nanoparticles and clusters generally involves a capping agent that provides control over their size, shape, composition, solubility, stability, chirality and functionality.2,10,14–17 The common capping agents employed in the synthesis are mainly amphiphilic surfactants, ionic liquids, polymers and organic ligands, which are carefully chosen depending on the application of interest.10,18,19 The metal–ligand interaction plays a crucial role in tuning the electronic structures of these particles, which influences their final properties, especially in catalysis.20,21 Hence, choosing an appropriate stabilising ligand is a crucial step while designing the synthesis procedure.
The catalytic activity of AuNPs is predominantly governed by geometric, electronic and steric factors. Geometric effects arise due to the orientation of Au atoms in space, and electronic effects originate from metal–ligand interactions or quantum size.10,22 Organic bound ligands can be used in the synthesis procedure, to enhance the electronic properties and impart extra stability to the AuNPs.21,23 The chemical and geometric environment around the gold atoms can be tuned by these bound ligands to modify the active sites and enhance the catalytic performance, similar to what is observed with enzymes.24,25 This strategy is robust, scalable and highly reliable for the size-selective synthesis of AuNPs. Therefore, this can be a useful approach to engineer novel functional materials at the nanoscale, according to the specific needs of the application considered.
In this study, organic ligand-bound gold nanoparticles are synthesised using a series of different gold(I) precursors (Fig. 1) in a one-step methodology. The different gold precursors are chloro(triphenylphosphine)gold(I) (P1), chloro(dimethylphenylphosphine)gold(I) (P2), and chloro(trimethylphosphine)gold(I) (P3). Their schematic structures are illustrated in Fig. 1a. These different gold(I) precursors lead to varying degrees of steric hindrance and are selected to study the role of the stabilising organic ligands and their effect on the final size of the synthesised AuNPs. As is evident from the chemical structures, phosphorus is surrounded by three phenyl rings in P1, which is highly sterically hindered, while, in P2, phosphorus is surrounded by only one phenyl and two methyl rings, and P3 is the least hindered with three methyl groups bound to the phosphorus atom. Fig. 1b shows the photographs of glass vials containing the AuNPs dissolved in ethanol immediately after the synthesis, using different precursors. AuNPs prepared from P1, P2, and P3 exhibit different colours: dark brown, pink, and purple, respectively. It is well known that the observed colour of the solution depends on the final size and shape of the AuNPs, because of surface plasmon resonance (SPR), a phenomenon that occurs due to collective oscillation of conduction electrons as induced by an electromagnetic field.26,27 In order to study the SPR and interaction of these particles with light, these solutions are analysed with UV/Vis spectroscopy. Characteristic peaks of SPR vary, depending upon the core diameter of AuNPs.27Fig. 1c represents the UV/Vis spectra of the gold nanoparticles synthesised using different precursors. The UV/Vis spectrum of Au-P1 consists of a broad band around 450 nm, which indicates that the diameters of the AuNPs are slightly larger than 1 nm. The UV/Vis spectrum of Au-P2 and Au-P3 shows SPR bands at 515 nm and 560 nm, respectively. The shift of the peak towards longer wavelengths (red shift) indicates that the size of the synthesised gold particles increases.28
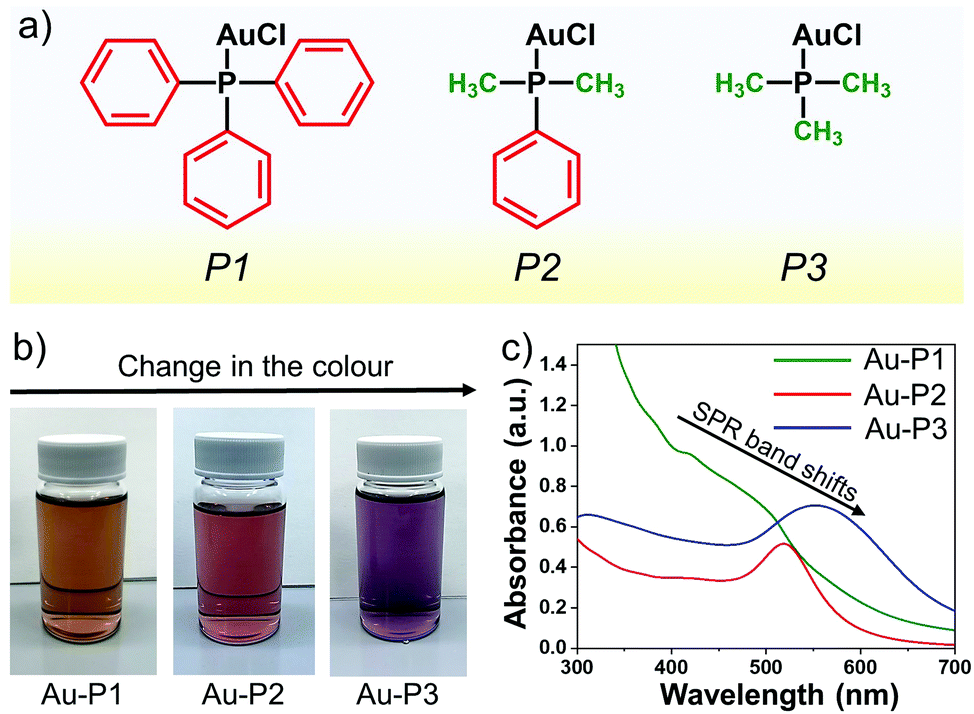 | ||
| Fig. 1 Synthesis and characterisation of gold nanoparticles (AuNPs) using different precursors. (a) Structures of different gold precursors used in this study. (b) Vials showing AuNPs in ethanol synthesised using different precursors. (c) UV/Vis spectra of Au-P1, Au-P2, and Au-P3 solutions. | ||
To confirm the final size of the particles, electron microscopy studies are performed on the samples; TEM images, along with corresponding particle size distributions are illustrated in Fig. 2a–f. The particle size of AuNPs synthesised from P1, P2 and P3 is found to be ∼1.5 ± 0.5 nm, 3.0 ± 1 nm and 9.8 ± 3 nm, respectively, which reveals that the degree of steric hindrance around the phosphorus atom plays an important role in steering the final size of the nanoparticles. Taken together, these data suggest that as the steric hindrance around the phosphorus atom decreases (P3), particles experience more freedom to aggregate and coalesce, forming larger particles that precipitate over time. The more hindered phosphorus (P1) assists in the stabilisation of the nanoparticles. Hence, a correlation is found relating the steric hindrance around phosphorus with the final size of the AuNPs. An ability to control and tune the size-dependent properties of the AuNPs according to the desired application is an important tool in materials synthesis.
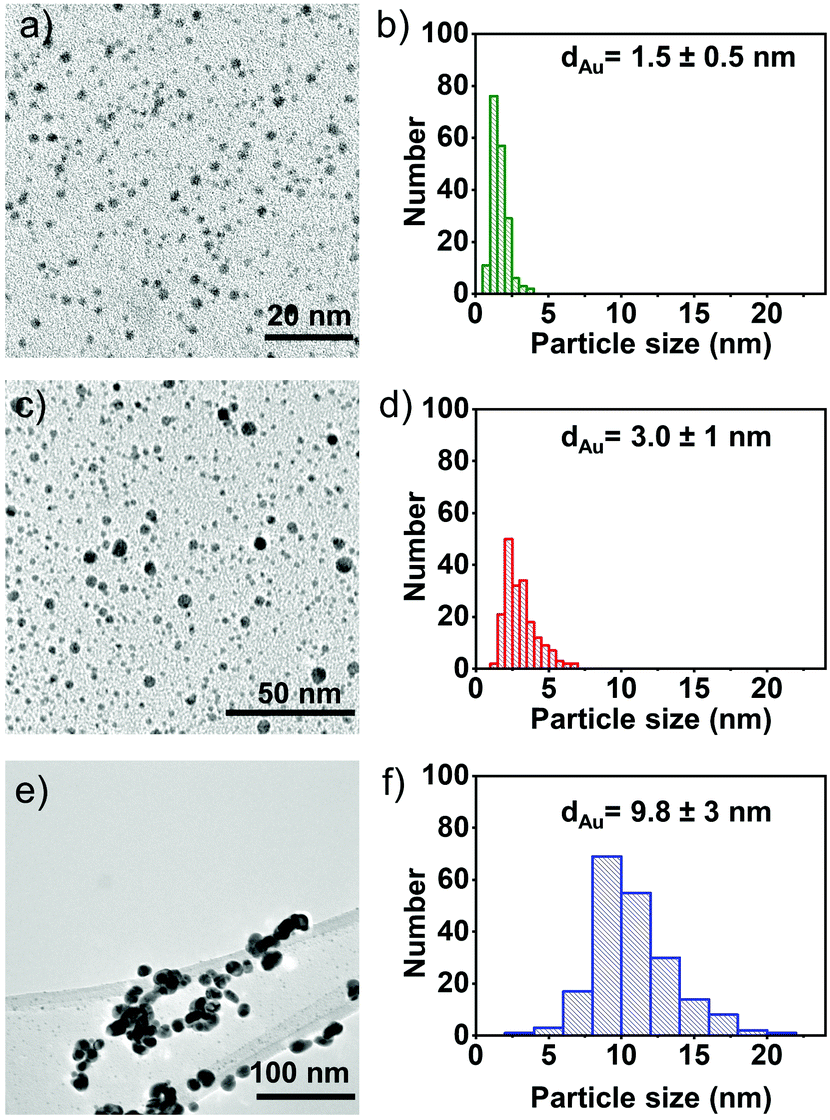 | ||
| Fig. 2 Bright-field TEM micrographs, along with the corresponding particle size distribution histogram of (a) and (b) Au-P1, (c) and (d) Au-P2 and (e), (f) Au-P3 solutions. | ||
Metallic nanoparticles are often deposited onto support materials to be utilised in different applications where liquid solutions are undesired, e.g., to be reused in heterogeneous catalysis.2,29 Here, we focus on propylene epoxidation as a catalytic application of these AuNPs. To further elucidate the effect of AuNPs size with varied ligands on the catalytic activity, the as-synthesised AuNP are directly immobilised on an untreated titanium silicalite-1 (TS-1) support (referred as Au-Pn/TS-1; Pn = precursor P1, P2 or P3; Au loading 0.5 wt%). Different coloured powders are obtained after immobilisation (Fig. 3a and b; inset). Direct immobilisation facilitates higher dispersion of Au and precise control over the particle size, which is critical for heterogeneous catalysis.21 The Au loading is determined using inductively coupled plasma optical emission spectroscopy (ICP-OES). The presence of Au on the TS-1 is confirmed using DR-UV/Vis spectroscopy and TEM. The DR-UV/Vis spectrum (ESI,† Fig. S6) consists of a surface plasmon band near 450 nm for Au-P1/TS-1, 540 nm for Au-P2/TS-1 and 550 nm for Au-P3/TS-1, confirming the presence of Au nanoparticles dispersed onto the TS-1 surface.30 The peak at around 350 nm reveals a small fraction of extra framework TiO2 and anatase species in the TS-1 sample.30 Furthermore, XRD patterns of these supported catalysts shown in ESI,† Fig. S7, confirm no change in the crystal structure of TS-1 after immobilisation. However, it was not possible to obtain clear Au peaks due to the small size of the particles.31 TEM images in Fig. 3a–c provide quantitative information on the final size of the clusters after immobilisation. Gold can be clearly seen on the external surface of TS-1, and the average particle size of the AuNPs is determined as ∼2.0 nm, 3.2 nm and 9.6 nm for Au-P1/TS-1, Au-P2/TS-1, and Au-P3/TS-1 after immobilisation, respectively. The bound ligands impart extra stability to the nanoparticles, but may lower the catalytic performance by blocking the access to the active sites and limiting reactant diffusion to the active sites.18,32 Therefore, it is necessary to remove the ligands before testing the catalytic activity of the AuNPs. Non-thermal O2 plasma treatment is employed for the removal of ligands from the supported catalyst.21,33 The removal of ligands and estimation of AuNP size is confirmed using thermogravimetric analysis (TGA), TEM imaging and DR-UV/Vis spectroscopy.
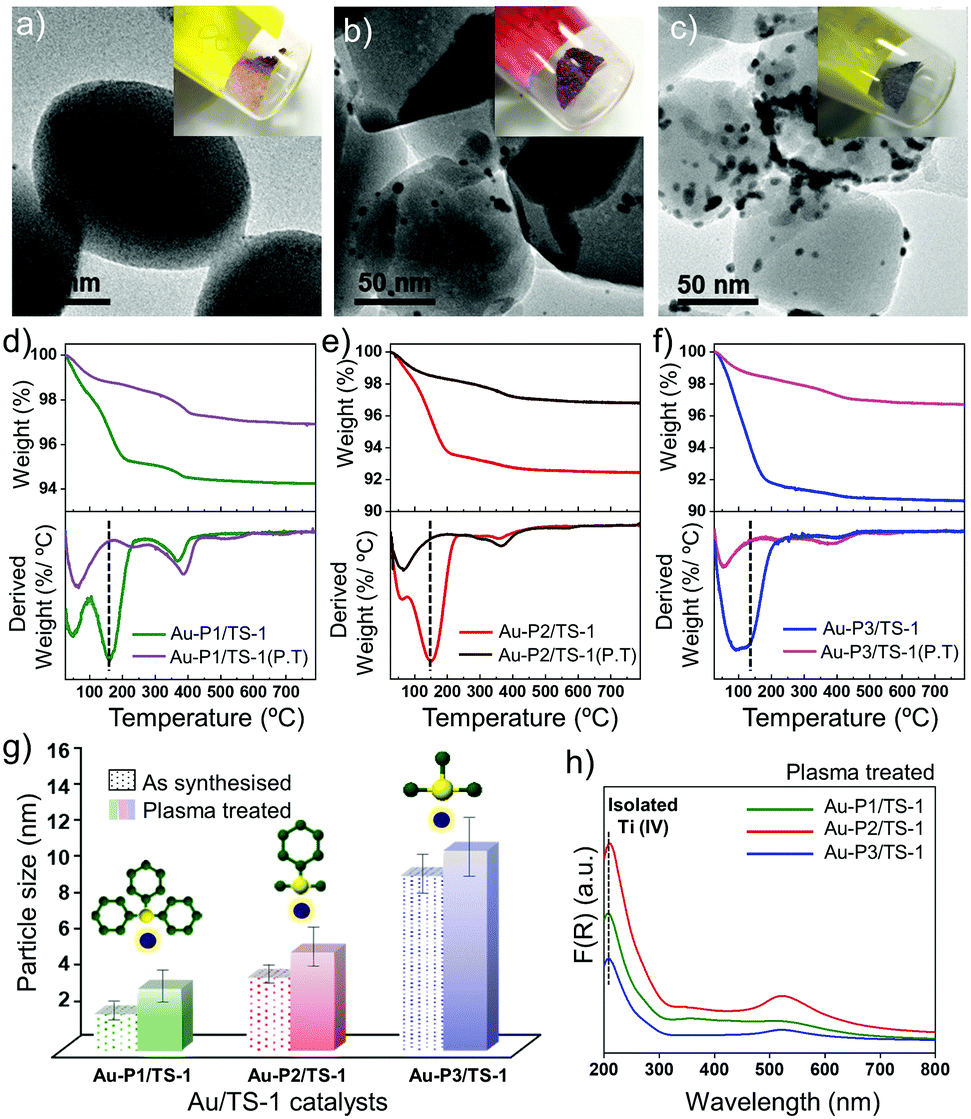 | ||
| Fig. 3 Synthesis and characterisation of differently sized AuNPs supported on TS-1. Representative TEM micrographs and a corresponding vial containing powder of (a) Au-P1/TS-1, (b) Au-P2/TS-1, (c) Au-P3/TS-1 catalysts. TGA and DTG analysis profile of (d) Au-P1/TS-1, (e) Au-P2/TS-1, (f) Au-P3/TS-1 catalysts before and after plasma treatment. (g) Bar graph showing different AuNP sizes calculated from TEM, before and after plasma treatment. (h) DR- UV/Vis spectra of Au/TS-1 catalysts after treating with plasma. | ||
Fig. 3d–f shows the TGA-DTG profiles of three different materials before and after the plasma treatment. From the data shown in the TGA profile, it can be calculated that 90% of the ligand is removed after the plasma treatment for each of the Au-P1/TS-1, Au-P2/TS-1, and Au-P3/TS-1 materials. This deduction is further supported by analysing the DTG profile. The broad peak at 100 °C in each curve is due to water loss, and the peak at around 200 °C corresponds to the organic bound ligands, which disappears in each material after the plasma treatment. TEM images of plasma treated Au-NP/TS-1 materials (ESI,† Fig. S8) reveal that there is a slight change in the final size of the AuNPs. The resulting AuNP size quantified from TEM are 3.4, 5.4 and 11 nm, synthesised using different precursors with decreasing steric hindrance around the phosphorus atom (P1 to P3, Fig. 3g). In addition to TGA and TEM, UV/Vis spectroscopy (Fig. 3h) shows that the peak at ∼ 450 nm for Au-P1/TS-1 shifts to ∼500 nm, indicating a particle size larger than 2 nm. The peaks for the Au-P2/TS-1 and Au-P3/TS-1 materials remain ∼520 nm. Along with that, the absorption peak at ∼208 nm, which is a characteristic of an isolated tetrahedrally coordinated Ti ion in the zeolite framework of TS-1, remains intact, suggesting no change in the zeolite structure. Taken cumulatively, these characterisations demonstrate that non-thermal plasma treatment facilitated the partial removal of these organic bound ligands from the material without affecting the catalyst morphology significantly.
To evaluate the size-dependent catalytic properties of supported AuNPs, the above synthesised materials Au-P1/TS-1, Au-P2/TS-1 and Au-P3/TS-1 (0.5 wt% Au) are tested for direct gas phase epoxidation of propylene using H2 and O2 in a fixed-bed quartz reactor at 225 °C. The samples exhibit stable catalytic behaviour, and results are illustrated in Fig. 4. Furthermore, we can establish a relationship between the AuNP size and the catalytic performance. The PO production rate of the Au-P1/TS-1 catalyst with the smallest particle size is observed to be 15 gPOh−1 kgcat−1 (propylene conversion ∼1.1%) with moderate PO selectivity of 78% (Fig. 4a and b). The Au-P2/TS-1 catalyst presented a slightly lower activity with a PO production rate of 12 gPOh−1 kgcat−1 (propylene conversion ∼0.9%) and similar PO selectivity of 78%. The PO production rate for Au-P3/TS-1 exhibits a very low PO production rate of 3.5 gPOh−1 kgcat−1 (propylene conversion ∼0.2%), accompanied by a low PO selectivity of 60%, implying that larger sized AuNPs lead to reduced PO formation. The PO formation rate and PO selectivity decrease remarkably as the Au particle size increases. This demonstrates that the smaller AuNPs have a more accessible area on their surface for the reaction, as compared to the larger AuNPs. The other by-products formed in this reaction are CO2, H2O, ethanal, and propanal. The H2 efficiency values for Au-P1/TS-1 slightly rise to 4.5% (ESI,† Fig. S9) and then gradually decrease to 3% as the time on-stream increases. The Au-P2/TS-1 and Au-P3/TS-1 display a steady H2 efficiency value of 3.8% and 2%, respectively. It is well known that highly dispersed small AuNPs, along with tetrahedrally coordinated Ti4+ sites located in the support, are required to generate in situ per-oxo species to produce PO.34 It can be clearly inferred from the above trends that the catalyst with the smallest particle size outperformed the catalyst with the largest particle size. In the case of large AuNPs, there are fewer Au atoms on the periphery, resulting in reduced proximity to Ti4+ sites, which increases the possibility of non-selective oxidation of propylene, resulting in a lower production of PO.35 Moreover, these large particles are active in hydrogen combustion to produce water, leading to low H2 utilisation and low PO selectivity.36 On the other hand, in smaller AuNPs, most of the Au atoms located on the surface are beneficial to form in situ peroxo species with Ti sites, capable of producing more PO and reducing the chance of other byproducts.37,38 Therefore, it can be confirmed that with decreasing AuNP size, the PO activity and selectivity increase. Table S1, ESI† displays a comparison of the catalytic performance of various Au on untreated TS-1 catalysts taken from the literature. Compared with the reported catalytic results, this Au-P1/TS-1 catalyst shows similar catalytic activity. However, overall propylene conversion and hydrogen efficiency of the synthesised catalysts is relatively low, which indicates that the Au–Ti interaction needs further improvement. The propylene conversion can be further increased by treating TS-1 with alkali metal promoters that has been reported to enhance Au–Ti interactions.39,40
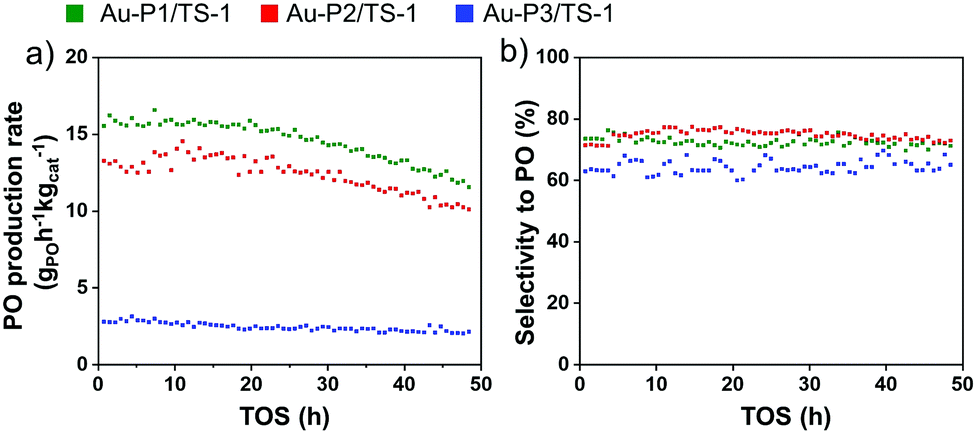 | ||
| Fig. 4 Catalytic performance of plasma treated Au-P1/TS-1, Au-P2/TS-1 and Au-P3/TS-1 catalysts. (a) Rate of PO production and (b) selectivity towards PO; C3H6/H2/O2/He = 1/1/1/7, Temperature: 225 °C, GHSV = 8000 mL gcat−1 h−1. | ||
The spent catalysts are analysed using TEM (ESI,† Fig. S10), and the TEM micrographs shown in the ESI† reveal polydispersity of nanoparticles after reaction for all the catalysts. The structure and the environment of the catalyst have a remarkable impact on the catalytic performance. The catalytic performance strongly depends on the size of the AuNP, which can be modulated by tuning the geometric environment and steric hindrance around the gold core.
In summary, a relationship is discovered between the steric hindrance around the phosphorus atom in the organic ligand and the final size of the AuNP. It is shown that as the steric hindrance around the phosphorus decreases in the Au+ precursor, larger particles are formed, and these are less stable over time. Furthermore, the pre-synthesised AuNPs are successfully immobilised onto untreated titanosilicalite-1 (TS-1) support, with very little change in the final size of the dispersed AuNPs. It is evident that Au-P1/TS-1, with the smallest gold particle size and highest stability in solution, shows the highest conversion and selectivity towards PO, compared to the other catalysts. Hence, a clear correlation between the final size of nanoparticles controlled by steric hindrance of ligands and catalytic activity is demonstrated. The insights provided here can be used to synthesise better catalysts through particle size control with organic ligands.
The authors gratefully acknowledge SABIC for financial support. The authors also acknowledge funding from EPSRC via ‘Frontier Engineering’ and ‘Frontier Engineering: Progression’ awards (grant numbers EP/K038656/1 and EP/S03305X/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- G. J. Hutchings, Catal. Today, 2005, 100, 55–61 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- N. Li, P. Zhao and D. Astruc, Angew. Chem., Int. Ed., 2014, 53, 1756–1789 CrossRef CAS PubMed.
- N. de Silva, J. M. Ha, A. Solovyov, M. M. Nigra, I. Ogino, S. W. Yeh, K. A. Durkin and A. Katz, Nat. Chem., 2010, 2, 1062–1068 CrossRef CAS PubMed.
- T. Ishida, T. Murayama, A. Taketoshi and M. Haruta, Chem. Rev., 2019, 120, 464–525 CrossRef PubMed.
- N. Lopez, T. Janssens, B. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232–235 CrossRef CAS.
- A. Corma, P. Concepción, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. López-Quintela and D. Buceta, Nat. Chem., 2013, 5, 775–781 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- G. J. Hutchings and M. Haruta, Appl. Catal., A, 2005, 291, 2–5 CrossRef CAS.
- M. M. Nigra, A. J. Yeh, A. Okrut, A. G. DiPasquale, S. W. Yeh, A. Solovyov and A. Katz, Dalton Trans., 2013, 42, 12762–12771 RSC.
- A. I. Kozlov, A. P. Kozlova, H. Liu and Y. Iwasawa, Appl. Catal., A, 1999, 182, 9–28 CrossRef CAS.
- G. Schmid, Chem. Soc. Rev., 2008, 37, 1909–1930 RSC.
- L. O. Brown and J. E. Hutchison, J. Am. Chem. Soc., 1999, 121, 882–883 CrossRef CAS.
- Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
- P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257, 638–665 CrossRef CAS.
- M. D. Wodrich and X. Hu, Nat. Rev. Chem., 2017, 2, 1–7 Search PubMed.
- N. Kapil, T. Weissenberger, F. Cardinale, P. Trogadas, T. A. Nijhuis, M. M. Nigra and M.-O. Coppens, Angew. Chem. Int. Ed., 2021, 60, 18185–18193 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- J.-M. Ha, A. Solovyov and A. Katz, Langmuir, 2009, 25, 10548–10553 CrossRef CAS PubMed.
- M.-O. Coppens, Curr. Opin. Chem. Eng., 2012, 1, 281–289 CrossRef CAS.
- P. Trogadas, M. M. Nigra and M.-O. Coppens, New J. Chem., 2016, 40, 4016–4026 RSC.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, J. Phys.: Condens. Matter, 2017, 29, 203002 CrossRef PubMed.
- B. Balamurugan and T. Maruyama, Appl. Phys. Lett., 2005, 87, 143105 CrossRef.
- M. Sankar, Q. He, R. V. Engel, M. A. Sainna, A. J. Logsdail, A. Roldan, D. J. Willock, N. Agarwal, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2020, 120, 3890–3938 CrossRef CAS PubMed.
- E. Sacaliuc, A. Beale, B. Weckhuysen and T. Nijhuis, J. Catal., 2007, 248, 235–248 CrossRef CAS.
- N. Li, Y. Chen, Q. Shen, B. Yang, M. Liu, L. Wei, W. Tian and J. Zhou, J. Solid State Chem., 2018, 261, 92–102 CrossRef CAS.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358–1362 CrossRef CAS.
- X. Liu, C.-Y. Mou, S. Lee, Y. Li, J. Secrest and B. W.-L. Jang, J. Catal., 2012, 285, 152–159 CrossRef CAS.
- J. Chen, S. J. A. Halin, E. A. Pidko, M. W. G. M. T. Verhoeven, D. M. P. Ferrandez, E. J. M. Hensen, J. C. Schouten and T. A. Nijhuis, ChemCatChem, 2013, 5, 467–478 CrossRef CAS.
- W.-S. Lee, L.-C. Lai, M. C. Akatay, E. A. Stach, F. H. Ribeiro and W. N. Delgass, J. Catal., 2012, 296, 31–42 CrossRef CAS.
- X. Feng, X. Duan, G. Qian, X. Zhou, D. Chen and W. Yuan, J. Catal., 2014, 317, 99–104 CrossRef CAS.
- D. H. Wells Jr, W. N. Delgass and K. T. Thomson, J. Catal., 2004, 225, 69–77 CrossRef.
- X. Feng, N. Sheng, Y. Liu, X. Chen, D. Chen, C. Yang and X. Zhou, ACS Catal., 2017, 7, 2668–2675 CrossRef CAS.
- W.-S. Lee, M. C. Akatay, E. A. Stach, F. H. Ribeiro and W. N. Delgass, J. Catal., 2013, 308, 98–113 CrossRef CAS.
- Z. Li, J. Zhang, D. Wang, W. Ma and Q. Zhong, J. Phys. Chem. C, 2017, 121, 25215–25222 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc04165g |
| This journal is © The Royal Society of Chemistry 2021 |