
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and development of a photoswitchable DFG-out kinase inhibitor†
Yongjin
Xu
a,
Chunxia
Gao
a,
Liliana
Håversen
b,
Thomas
Lundbäck
 c,
Joakim
Andréasson
*d and
Morten
Grøtli
*a
c,
Joakim
Andréasson
*d and
Morten
Grøtli
*a
aDepartment of Chemistry and Molecular Biology, University of Gothenburg, SE-41296 Gothenburg, Sweden. E-mail: grotli@chem.gu.se
bDepartment of Molecular and Clinical Medicine, Institute of Medicine, Sahlgrenska Academy at University of Gothenburg, SE-41345 Gothenburg, Sweden
cMechanistic & Structural Biology, Discovery Sciences, R&D, AstraZeneca, SE-48183 Mölndal, Sweden
dDepartment of Chemistry and Chemical Engineering, Physical Chemistry, Chalmers University of Technology, SE-41296 Gothenburg, Sweden. E-mail: a-son@chalmers.se
First published on 7th September 2021
Abstract
We report the synthesis and characterisation of a photoswitchable DFG-out kinase inhibitor. Photocontrol of the target kinase in both enzymatic and living cell assays is demonstrated.
Protein kinases play important roles in signal transduction cascades, regulating different cellular processes such as cell differentiation, cell proliferation, and apoptosis. Thus, dysregulation of key protein kinases often leads to severe disorders, including cancer, inflammation, and neuro-degenerative diseases.1 Furthermore, the role that many kinases play in a variety of disease states is poorly understood. Reversibility and temporal control over kinase inhibition will contribute to a better understanding of kinase function in biological systems. In addition, the kinase of interest may have a specific function in an organ or tissue. The ability to regulate the kinase activity in a site-specific manner will have positive implications in proving the site of action of the kinase inhibitor, studying cell signalling, and understanding adverse effects of the inhibitor.
Photopharmacology has become a term for a number of approaches in which photochemistry and pharmacology are linked to achieve efficient spatiotemporal control over the biological activities of small molecules by light.2 One of the most widely used approaches to modify bioactive molecules into light-responsive counterparts is integration with molecular photoswitches. Such photoswitchable structures can be reversibly isomerised by light at different wavelengths. More recently, this approach has emerged as a powerful strategy to control a range of different targets, including receptors and ion channels,2a,3 lipid membranes,3 enzymes,2a,3 and nucleic acids.2a,4 So far, only a few photoswitchable molecules have been reported to target kinases, such as RET and BRAFV600E (Fig. 1A).5–10 The development of these compounds has been challenging because of common photopharmacology issues,10–12 such as insufficient isomeric enrichment in the photo-stationary distributions (PSDs); narrow bioactive discrepancy between the two isomers; rapid thermal isomerization; and reduction by, e.g., dithiothreitol (DTT) and glutathione (GSH), which are typically used for protein stabilisation in biological assays.
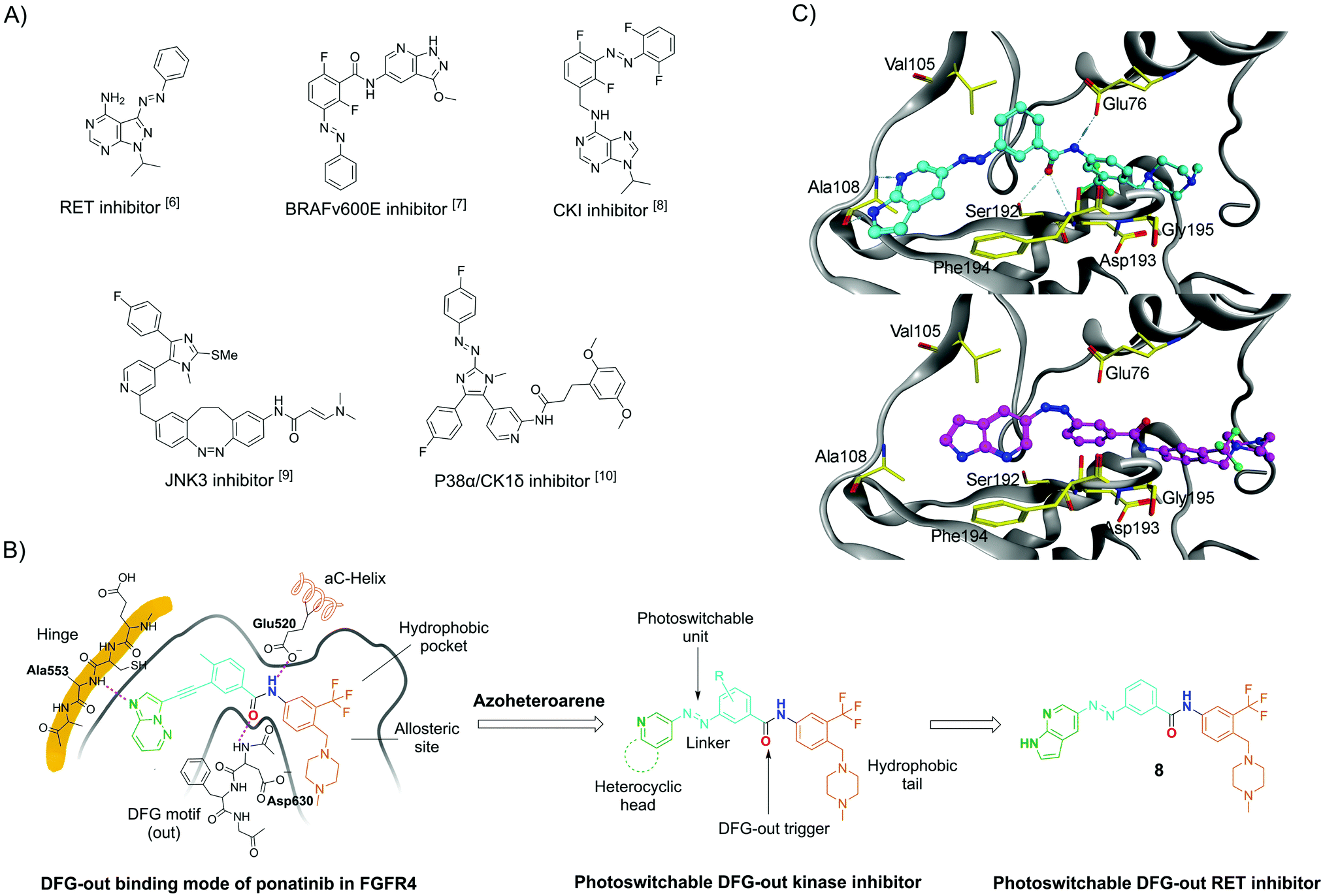 | ||
| Fig. 1 (A) Selected azo-based photoswitchable kinase inhibitors reported in the literature; (B) DFG-out binding model of ponatinib in FGFR4 kinase (PDB code 4UXQ), which is highly homologous to the RET kinase, inspired the design of the herein-reported photoswitchable DFG-out RET kinase inhibitor 8. (C) Computational modelling of 8E (top) and 8Z (bottom) in the DFG-out RET kinase. The DFG-out model was obtained by homology modelling, which applied the RET protein sequence (NCBI database GeneID: 547807) on a template of KIT crystal structure (PDB code 4U0I).22 | ||
REarranged during Transfection (RET) is a kinase belonging to the receptor tyrosine kinase family. It is essential for the development neurons, and dysregulation of RET activity is an important contributor to several human cancers.13 Small molecule inhibitors against RET have been developed as potential anti-cancer agents.14 Herein, we report the synthesis, photophysical characterisation, and biological evaluation of a photoswitchable DFG-out kinase inhibitor, using RET as a model target.
DFG-out kinase inhibitors (also named type II inhibitors) represent a class of bioactive molecules that trap their target kinases in an inactive, so-called DFG-out state, occupying a hydrophobic pocket adjacent to the ATP binding site.15 Ponatinib is a DFG-out kinase inhibitor with potent activity toward wild-type and drug-resistant gatekeeper mutations in the RET kinase domain.16,17 Using ponatinib as the template, we designed a potential photoswitchable DFG-out RET kinase inhibitor (compound 8; see Fig. 1B for structure). By replacing the acetylene moiety of ponatinib with an E-form azo bridge, the key interactions with the adenine-binding pocket and the geometry for triggering DFG-out conformation are preserved. Conversely, we hypothesized that the RET kinase adenine-binding pocket would not tolerate the inhibitor in the Z-form, thus weakening or even losing the inhibition effect. This would result in an inhibitor in which the stable isomer (E) is more potent. In principle, this is a drawback, as it causes the molecule to be constitutively active in its thermally stable state.18 This disadvantage is small when the Z-isomer can be enriched almost quantitative and has very good thermal stability. One way to circumvent this drawback would be to use the “sign inversion” strategy. However, this strategy relies on other photoswitches, such as hemithioindigos and diazocines, and these photoswitches could not be incorporated into the pharmacophore of the DFG-out inhibitor (data not shown). We decided to use 7-azaindole as a “head” group because it has been found to be an excellent hinge-binding motif, making two hydrogen bonds with the kinase hinge region.19,20 Furthermore, it had been reported that 7-azaindole-based azoheteroarene possesses excellent photophysical properties.21 To further evaluate our design, compounds 8E and 8Z were docked with the RET kinase (see Fig. 1C). For this purpose, homology modelling of the DFG-out RET structure was carried out using KIT kinase as a template structure (PDB code 4U0I). For the E-isomer, the 7-azaindole moiety occupies approximately the same space as the heteroaromatic ring of ponatinib, allowing for two hydrogen bonds with the kinase hinge region. The E-isomer azo bridge linkage directs the remaining portion of the molecule deep into the rear corner of the ATP-binding pocket. Here, the phenyl ring traverses the region adjacent to the kinase gatekeeper residue (Val105), and the 3-trifluoromethylphenyl aromatic ring engages the hydrophobic pocket in the allosteric site. Importantly, the amide bond stabilizes the outward flipping of the DFG motif. However, docking experiments with the Z-isomer shows that the key interactions with the hinge and the stabilization of the DFG-out cannot be maintained.
Scheme 1 shows the synthetic route of target compound 8. The aza-bridge containing moiety was prepared from 1 by an Ullmann-type coupling, followed by a Buchwald–Hartwig coupling with 3 to connect the heterocyclic head and the linker to form the key intermediate 4.21 Subsequent Boc-deprotection, oxidation, and removal of the tosyl group and hydrolysis of the ester furnished 6. Finally, coupling the “tail” part 723 to 6 yielded the target compound 8. The HCl salt form of 8 was prepared by treating the compound with 4 N HCl in 1,4-dioxane and evaporating the solvent.
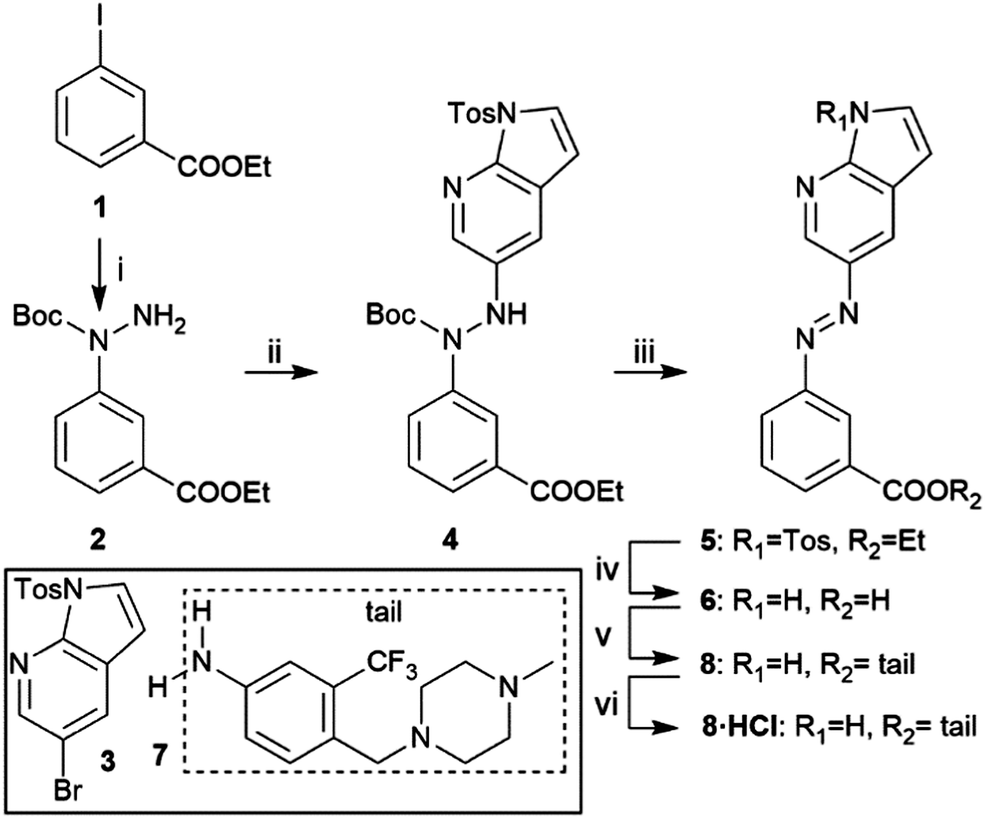 | ||
| Scheme 1 Synthesis of compound 8. Reagents and conditions: (i) Boc-hydrazide, CuI, Cs2CO3, DMSO, 50 °C, 4 h, 85%; (ii) 3, Pd(OAc)2, Xantphos, Cs2CO3, toluene, MW, 110 °C, 2 h, 86%; (iii) DMF, MW, 180 °C, 40 min; then O2, 100 °C, 2 h, 56%; (iv) NaOH, EtOH/H2O, 90 °C, 5 h, 92%; (v) 7, EDC·HCl, HOBt, Et3N, DCM, r.t., 36 h, 78%; (vi) 4 N HCl in 1,4-dioxane, quantitative. | ||
With the target azo compound 8 and its salt form 8·HCl in hand, their photophysical properties were studied in DMSO and aqueous solution, respectively. As for the DMSO solution, a thermally adapted sample (>99% in the E-isomer) was first irradiated by UV light (λ = 365 nm, 14 mW cm−2 for 15 seconds) to reach a containing 97% Z-isomer (Fig. 2A) as judged by the 1H-NMR spectrum (Fig. S8, ESI†). This UV-enriched sample was subsequently exposed to blue light (λ = 460 nm, 12 mW cm−2 for 20 seconds), yielding 64% E-isomer. The UV-vis absorption spectra of 8·HCl in H2O was recorded analogously as for 8 in DMSO (Fig. 2B). It is worth noticing that both samples show excellent fatigue resistance, as no significant degradation was observed after 10 cycles of UV-vis switching (Fig. 2A and B, insets). The thermal stability of 8Z is very good in both DMSO and aqueous solution, displaying a thermal half-life of 377 h and 629 h at room temperature, respectively (Fig. 2C and D). Thus, the photophysical properties of 8 in DMSO and in aqueous solution are comparable.
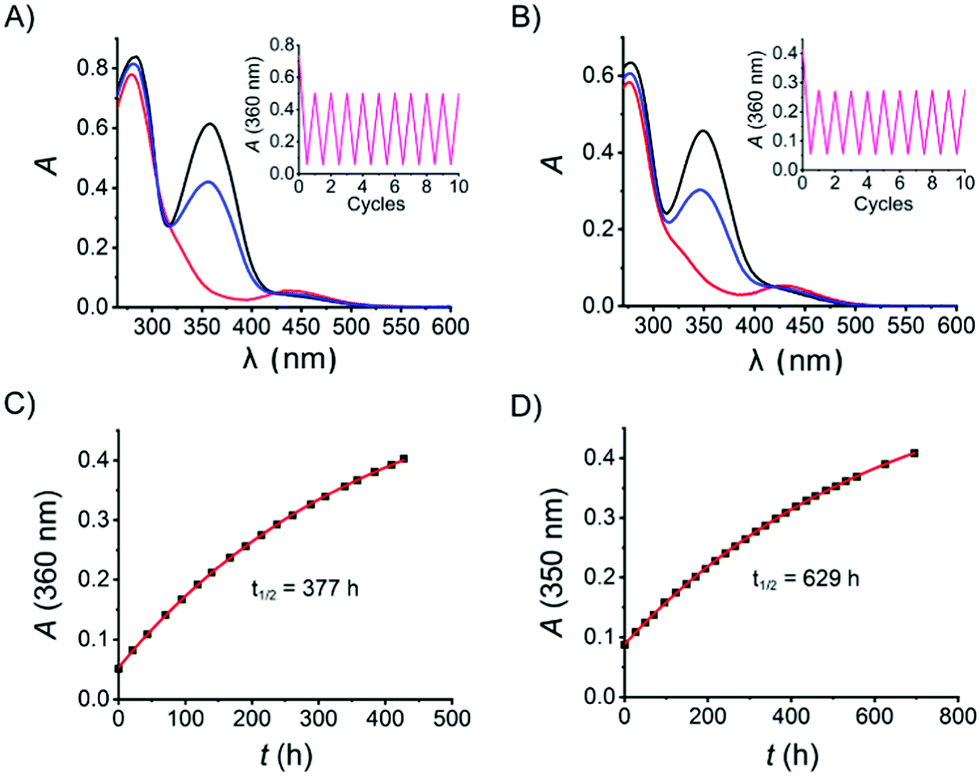 | ||
| Fig. 2 Photoisomerization UV-vis absorption spectrum of 8 as DMSO solution (25 µM, A) and 8·HCl in aqueous solution (25 µM, B): black line: at thermal equilibrium. Red line: after exposure to 365 nm UV to yield the PSD. Blue line: after exposure to 460 nm light to yield the PSD. The insets show the absorption changes during 10 cycles of UV and vis exposure (A and B). Thermal isomerization of the Z-isomer to the E-isomer monitored by UV-vis absorption of 8 in DMSO (C) and 8·HCl in H2O (D). | ||
Reductive degradation is a potentially limiting factor for the use of azobenzene photoswitches in biology,10,24 while DTT is a reducing reagent commonly used in biochemical assays to keep the proteins in a reduced state, and GSH is a natural antioxidant in mammalian cells. To ensure that the herein-reported azo compound is stable over DTT/GSH incubation during the biological assay described below, compound 8 was incubated with DTT (2 mM) and GSH (10 mM) in aqueous solution, respectively, and the stability was monitored by LC-MS. No degradation products were detected after 18 h incubation at room temperature (see ESI†). Subsequent UV-vis cyclicisomerisation of 8·HCl in aqueous solution containing 1 mM GSH was performed, resulting in no detectable degradation (Fig. 3A).
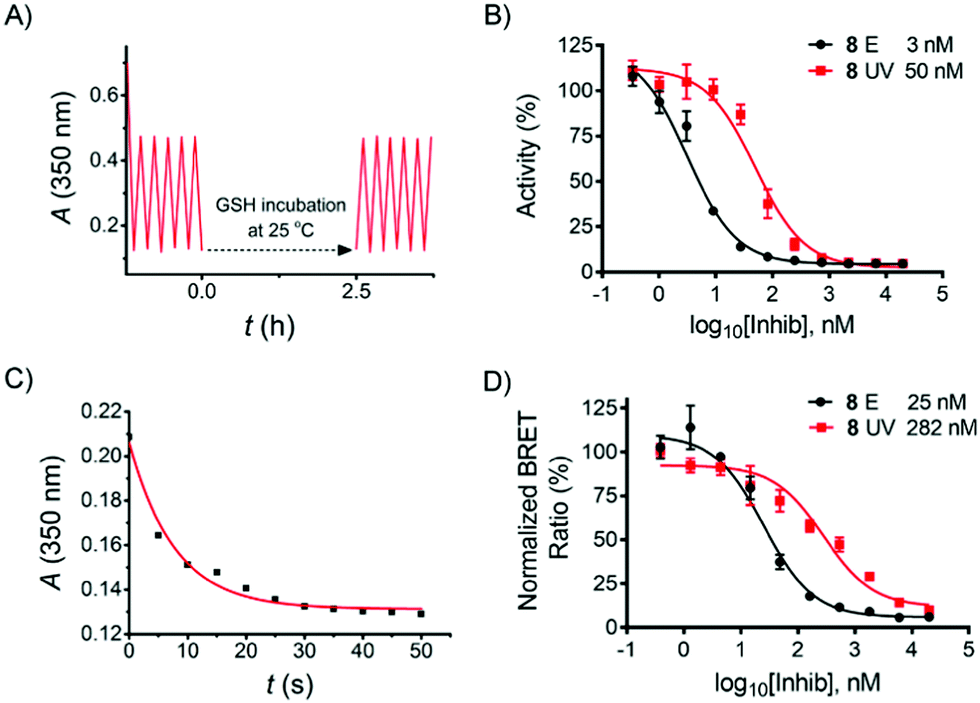 | ||
| Fig. 3 (A) GSH reduction assay, 8 (35 µM) in H2O with 1% DMSO and 1 mM GSH; (B) inhibitory concentration-response curve of E- and Z-isomer of 8 on RET activity as analysed by ADP-GloTM RET Kinase Assay; (C) photoswitching kinetics of 8 in cell assay medium (20 µM) (D) concentration-response curve of E- and Z-isomer of 8 as determined using NanoBRET™ TE Intracellular RET Kinase Assay in HEK293 cells transfected with RET-Luciferase. The data in (B) and (D) represent means from triplicate experiments. | ||
In order to evaluate the inhibitory effect of 8, the compound was tested in a biochemical ADP-Glo™ RET Kinase Assay. As type II inhibitors are reported to be slow binders,25,26 a pre-incubation step was added to the assay protocol (see ESI†). In addition, 8 in DMSO was thermally adapted to the pure E-isomer by heating prior to the assay, while the Z-isomer was prepared by irradiating the serially diluted DMSO solution in a 96-well plate (4 µl in each well) with a 365 nm LED array for 15 min prior to further dilution by kinase reaction buffer. All kinase reactions were performed at 10 µM ATP concentration. As a result, compound 8E (IC50 = 3 nM) exhibited 17-fold more potent activity than 8Z (IC50 = 50 nM) (Fig. 3B). The inhibitory effect of 8E is comparable with the reference compounds (staurosporine IC50 = 8 nM; ponatinib IC50 = 3 nM; see ESI,† Section 4.3).
Encouraged by these results, compound 8 was further evaluated in a NanoBRET™ TE Intracellular Kinase Assay. This assay allows evaluating compounds that bind to a kinase protein fused to NanoLuc luciferase in a competitive format by using a cell-permeable fluorescent NanoBRET tracer in HEK293 cells (human embryonic kidney cells). As shown in Fig. 3D, the IC50 of the E-isomer was determined to be 25 nM (incubation at 37 °C for 90 min). To evaluate the activity of the Z-isomer, 8E was initially incubated, followed by stepwise in situ irradiation to maximize the UV-induced PSD of the Z-isomer throughout the incubation (see ESI,† Section 5). This irradiation procedure was performed due to the relatively long residence time of the E-isomer bound in the kinase pocket when irradiation was performed,23 and it is mostly assumed that ligand photoswitching occurs only while the ligand is not bound to its target.2b In addition, as shown in Fig. 3C, 35 s of UV exposure is sufficient to switch the E-isomer to the Z-isomer-enriched PSD in cell assay medium. Therefore, a total of 35 s of UV light was delivered to the cells in three steps (15 s, 10 s, 10 s). An IC50 of the Z-isomer-enriched sample of 282 nM resulted (Fig. 3D), i.e., a factor of 11 times higher than that observed for the pure E-isomer.
In summary, we have designed and developed a practical photoswitchable DFG-out RET kinase inhibitor derived from ponatinib. This molecule shows efficient photoswitching to generate a nearly pure Z-isomer in the UV-induced PSD, as well as excellent thermal stability at room temperature. The E-isomer potently inhibits the RET kinase with single-digit nM IC50 in an enzymatic assay, while the UV-enriched Z-isomer exhibited 17 times less inhibition activity. More importantly, we can regulate the activity of the inhibitor in a cellular context by in situ irradiation, which shows an 11-fold activity difference between the assays with pure E-isomer and UV-enriched Z-isomer, while the E-isomer still maintains high potency. The almost quantitative switching of the E- to the Z-isomer combined with the very high thermal stability of 8Z set the stage for incubating cells with the inactive conformation and after reaching the target site the compound can be turned “on” with light.18 The successful in vitro application paves the way for further in vivo applications, e.g., in zebrafish. Furthermore, a significant fraction of the kinome is likely targetable by type II inhibitors,27 and this work will pave the way for developing photoswitchable DFG-out inhibitors targeting other kinases.
Conceptualisation and supervision; JA and MG. Investigation and methodology; all authors. Writing – original draft; YX. Writing – review and editing: YX, MG, JA. Funding acquisition, project administration and resources; JA and MG.
The authors acknowledge financial support from the Swedish Research Council (grants no. 2016-0360 to J. A., 2015-05642 to M. G. and 2019-03991 to M. G.). We also thank Dr Simon Moussaud (CBCS) for help with the biological assays.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900 CrossRef CAS; (b) J. Broichhagen, J. A. Frank and D. Trauner, Acc. Chem. Res., 2015, 48, 1947 CrossRef CAS.
- (a) Q. Liu and A. Deiters, Acc. Chem. Res., 2014, 47, 45 CrossRef CAS PubMed; (b) A. S. Lubbe, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2017, 46, 1052 RSC.
- D. Wilson, J. W. Li and N. R. Branda, ChemMedChem, 2017, 12, 284 CrossRef CAS PubMed.
- R. Ferreira, J. R. Nilsson, C. Solano, J. Andréasson and M. Grøtli, Sci. Rep., 2015, 5, 9769 CrossRef CAS PubMed.
- M. Hoorens, M. E. Ourailidou, T. Rodat, P. E. Wouden, P. Kobauri, M. Kriegs, C. Peifer, B. L. Feringa, F. J. Dekker and W. Szymanski, Eur. J. Med. Chem., 2019, 179, 133 CrossRef CAS PubMed.
- R. Ferreira, J. R. Nilsson, C. Solano, J. Andréasson and M. Grøtli, Sci. Rep., 2015, 5, 9769 CrossRef CAS PubMed.
- M. Hoorens, M. E. Ourailidou, T. Rodat, P. E. Wouden, P. Kobauri, M. Kriegs, C. Peifer, B. L. Feringa, F. J. Dekker and W. Szymanski, Eur. J. Med. Chem., 2019, 179, 133 CrossRef CAS PubMed.
- D. Kolarski, C. Miró-Vinyals, A. O. Sugiyama, A. Srivastava, D. Ono, Y. Nagai, M. Iida, K. Itami, F. Tama, W. Szymanski, T. Hirota and B. L. Feringa, Nat. Commun., 2021, 12, 3164 CrossRef CAS.
- M. Reynders, A. Chaikuad, B.-T. Berger, K. Bauer, P. Koch, S. Laufer, S. Knapp and D. Trauner, Angew. Chem., Int. Ed., 2021, 60, 20178 CrossRef CAS PubMed.
- M. Schehr, C. Ianes, J. Weisner, L. Heintze, M. P. Müller, C. Pichlo, J. Charl, E. Brunstein, J. Ewert, M. Lehr, U. Baumann, D. Rauh, U. Knippschild, C. Peifer and R. Herges, Photochem. Photobiol. Sci., 2019, 18, 1398 CrossRef CAS PubMed.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436 CrossRef CAS PubMed.
- D. Kolarski, A. Sugiyama, T. Rodat, A. Schulte, C. Peifer, K. Itami, T. Hirota, B. L. Feringa and W. Szymanski, Org. Biomol. Chem., 2021, 19, 2312 RSC.
- L. M. Mulligan, Nat. Rev. Cancer, 2014, 14, 173 CrossRef CAS PubMed.
- L. Mendoza, Oncol. Rev., 2018, 12, 352 Search PubMed.
- Y. Liu and N. S. Gray, Nat. Chem. Biol., 2006, 2, 358 CrossRef CAS PubMed.
- D. Plenker, M. Riedel, J. Brägelmann, M. A. Dammert, R. Chauhan, P. P. Knowles, C. Lorenz, M. Keul, M. Bührmann and O. Pagel, et al. , Sci. Transl. Med., 2017, 9, 6144 CrossRef PubMed.
- L. Mologni, S. Redaelli, A. Morandi, I. Plaza-Menacho and C. Gambacorti-Passerini, Mol. Cell. Endocrinol., 2013, 377, 1 CrossRef CAS.
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymański, Chem. Sci., 2020, 11, 11672 RSC.
- T. Irie and M. Sawa, Chem. Pharm. Bull., 2018, 66, 29 CrossRef CAS PubMed.
- B. Daydé-Cazals, B. Fauvel, M. Singer, C. Feneyrolles, B. Bestgen, F. Gassiot, A. Spenlinhauer, P. Warnault, N. Van Hijfte, N. Borjini, G. Chevé and A. Yasri, J. Med. Chem., 2016, 59, 3886 CrossRef PubMed.
- Y. Xu, C. Gao, J. Andréasson and M. Grøtli, Org. Lett., 2018, 20, 4875 CrossRef CAS PubMed.
- J. A. Tucker, T. Klein, J. Breed, A. L. Breeze, R. Overman, C. Phillips and R. A. Norman, Structure, 2014, 22, 1764 CrossRef CAS PubMed.
- W. Shen, M. S. Tremblay, V. A. Deshmukh, W. Wang, C. M. Filippi, G. Harb, Y. Zhang, A. Kamireddy, J. E. Baaten and Q. Jin, et al. , J. Am. Chem. Soc., 2013, 135, 1669 CrossRef CAS PubMed.
- C. Boulegue, M. Loeweneck, C. Renner and L. Moroder, ChemBioChem, 2007, 8, 591 CrossRef CAS PubMed.
- J. Regan, C. A. Pargellis, P. F. Cirillo, T. Gilmore, E. R. Hickey, G. W. Peet, A. Proto, A. Swinamera and N. Moss, Bioorg. Med. Chem. Lett., 2003, 133, 3101 CrossRef.
- C. Pargellis, L. Tong, L. Churchill, P. F. Cirillo, T. Gilmore, A. G. Graham, P. M. Grob, E. R. Hickey, N. Moss, S. Pav and J. Regan, Nat. Struct. Biol., 2002, 9, 268 CrossRef CAS PubMed.
- Z. Zhao, H. Wu, L. Wang, Y. Liu, S. Knapp, Q. Liu and N. S. Gray, ACS Chem. Biol., 2015, 9, 1230 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedure and characterization of compounds, NMR spectra, HRMS spectra, photophysical characterization, biological characterization and protocol for docking. See DOI: 10.1039/d1cc04125h |
| This journal is © The Royal Society of Chemistry 2021 |