
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriggering a transient organo-gelation system in a chemically active solvent†
Romain
Chevigny‡
 a,
Johanna
Schirmer‡
a,
Carmen C.
Piras
b,
Andreas
Johansson
ac,
Elina
Kalenius
a,
David K.
Smith
b,
Mika
Pettersson
a,
Efstratios D.
Sitsanidis
*a and
Maija
Nissinen
*a
a,
Johanna
Schirmer‡
a,
Carmen C.
Piras
b,
Andreas
Johansson
ac,
Elina
Kalenius
a,
David K.
Smith
b,
Mika
Pettersson
a,
Efstratios D.
Sitsanidis
*a and
Maija
Nissinen
*a
aDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, P.O. Box 35, FI-40014 JYU, Finland. E-mail: maija.nissinen@jyu.fi
bDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK
cDepartment of Physics, Nanoscience Center, University of Jyväskylä, P.O. Box 35, FI-40014 JYU, Finland
First published on 15th September 2021
Abstract
A transient organo-gelation system with spatiotemporal dynamic properties is described. Here, the solvent actively controls a complex set of equilibria that underpin the dynamic assembly event. The observed metastability is due to the in situ formation of a secondary solvent, acting as an antagonist against the primary solvent of the organogel.
In nature, energy dissipating biomolecular assemblies form transiently under the constant exchange of energy and matter with the environment. However, many artificial assembled systems lack the complexity of their natural counterparts as the assembly event occurs at thermodynamic equilibrium. There is increasingly intense interest in metastable gelation systems that assemble in a dynamic, kinetically controlled way, for example, in response to the presence of fuel and then disassemble when the fuel is depleted.1–3 Van Esch et al., for example, used an alkylating agent as fuel to generate a low molecular weight gelator (LMWG) ester, while the process was reversed by ambient hydrolysis.4 A number of reports built on this demonstrating that hydrogels can exist in non-equilibrium states using various fuels.5–8
Transient materials do not only depend on cyclic fuelled dissipative systems. In the seminal work of Heuser et al.,9 the catalytic temporal control of a pH-responsive dipeptide consists of an alternative approach towards transient peptide gels. The self-assembly of LMWGs is a dynamic process, strongly dependent on their structural features and the nature of the gelation trigger, either physical or chemical.10,11 For example, photoresponsive gelators have been reported to assemble towards out-of-equilibrium systems,12,13 deprotection of a Boc group has been used to convert organogelators to hydrogelators,14 simple deprotection reactions can yield gels in situ,15 and enzyme-mediated chemical changes can trigger self-assembly.16–18 Catalysis plays an intimate role in controlling non-equilibrium fuelled systems in both assembly/disassembly pathways. A case as such describes gelation in which the fuel derives catalytically from a “pre-fuel”.19 Additionally, catalysis has been reported to trigger an interplay between pH and oxidation, leading to the transient assembly of dendritic peptide monomers.20 Among amino acid based gelators, phenylalanine derivatives are efficient LMWGs, yielding self-supporting gels.21,22 Working on a multistep synthesis towards a set of phenylalanine based organogelators, we serendipitously noticed the in situ gelation of dipeptide N-tert-butyloxycarbonyl (Boc)–L-phenylalanyl–L-phenylalanine tert-butyl ester (Boc–Phe–Phe–OtBu 1), triggered by the selective deprotection of the Boc group in the presence of the tert-butyl group (Fig. 1 and Fig. S1, ESI†).
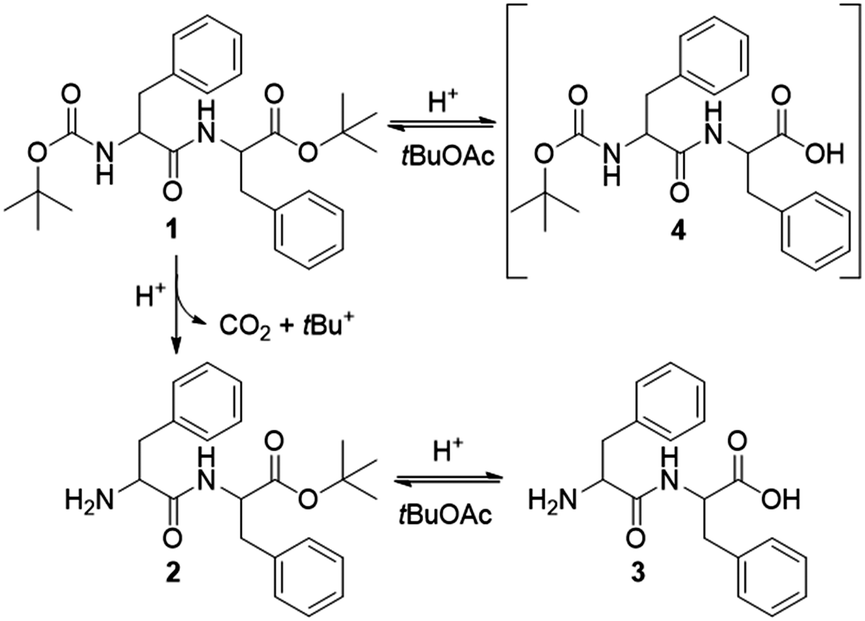 | ||
| Fig. 1 Selective deprotection of the Boc-group in the presence of the tert-butyl group under acidic conditions. Gelation is induced by the chemical equilibrium between products 2 and 3. | ||
Herein, we focus on deciphering the gelation mechanism and understanding how the dynamic nature of the reaction equilibrium affects the stability of the obtained organogel. In many cases, the solvent in gelation systems plays a relatively passive role in simply supporting the assembly event. In this system, however, it plays an active and chemically intimate role in controlling the equilibria that underpin the dynamic assembly process.
The deprotection reaction is based on the protocol of Lin et al.23 The precursor gelator 1 is suspended in tert-butyl acetate (tBuOAc) at a concentration of 0.5 M. When adding 1.5 equivalents of sulfuric acid to the reaction mixture, dipeptide 1 is selectively deprotected at the carbamate group to give the ester Phe–Phe–OtBu 2 (Fig. 1 and S2, ESI†) in 95% yield. According to Lin et al.,23 the driving force of the reaction is the formation and release of carbon dioxide formed by the irreversible cleavage of the Boc group. Their studies showed that the C-terminal ester is also deprotected to give the mono-carbamate protected dipeptide Boc–Phe–Phe 4. This process is rapid and reversible as tBuOAc regenerates the tertiary cation (tBu+), therefore reforming the ester group of 1. In this way, the solvent plays an intimate role in the reaction cycle. However, our studies showed that gelation is achieved when adding less sulfuric acid (1.0 equivalent) while keeping the concentration of 1 constant (0.5 M). In this sense, sulfuric acid can be considered an “accelerator” for the gelation process.
To identify the minimum gelation concentration (MGC) of precursor 1 (0.05 M; Table S1, ESI†), we performed a series of concentration screening trials. Gelation, assessed by the vial inversion method, occurred within 12 h upon addition of the acid at room temperature. In addition, we measured the gel-to-sol phase transition temperature (Tgel–sol) by controlled heating of the gels. Irrespective of their concentration, all samples appeared stable up to 45–50 °C. Heating at 55 °C resulted in the collapse of the gels to a solution. However, they reformed upon cooling, verifying the thermoreversible nature of the gel (Fig. S3, ESI†).
The organogel has a lifespan of four days, after which it turns into a solution. Under acidic conditions, the deprotection of 1 could occur to a full extent towards L-phenylalanyl–L-phenylalanine (Phe–Phe) 3 before tBuOAc regenerates ester 2. Therefore, gelation must be triggered either by Phe–Phe 3, the mono-protected ester 2 or both potential gelators 2 and 3. The slow decay of the organogel within four days could suggest possible changes in the chemical equilibrium between dipeptide counterparts 2 and 3. Therefore, to identify the gelling agents and investigate the gelation mechanism with respect to the reaction equilibrium, we performed nuclear magnetic resonance (NMR) studies on the dried gel (xerogel I-dried xerogel dissolved in deuterated d6-DMSO) of a freshly prepared organogel.
Both 13C and 1H NMR spectra of xerogel I confirmed the presence of the mono-protected 2 and fully deprotected 3 dipeptides, in approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.9 ratio (Fig. 2 and Fig. S5, S6, ESI†). The two doublets at ∼9.0 ppm are attributed to the two NH groups of the peptide bonds. Integration of the remaining signals shows twice the number of H atoms, except for the tert-butyl group. The broad band at 4.4 ppm is due to the hydroxyl group of tert-butyl alcohol (tBuOH), a side product of the ester 2 deprotection. The broad peak at ∼8 ppm corresponds to the protonated N-terminus of compounds 2 and 3 under acidic conditions. The presence of both compounds in the xerogel was also confirmed by high-resolution mass spectroscopy (HR-MS, Fig. S10, ESI†).
:0.9 ratio (Fig. 2 and Fig. S5, S6, ESI†). The two doublets at ∼9.0 ppm are attributed to the two NH groups of the peptide bonds. Integration of the remaining signals shows twice the number of H atoms, except for the tert-butyl group. The broad band at 4.4 ppm is due to the hydroxyl group of tert-butyl alcohol (tBuOH), a side product of the ester 2 deprotection. The broad peak at ∼8 ppm corresponds to the protonated N-terminus of compounds 2 and 3 under acidic conditions. The presence of both compounds in the xerogel was also confirmed by high-resolution mass spectroscopy (HR-MS, Fig. S10, ESI†).
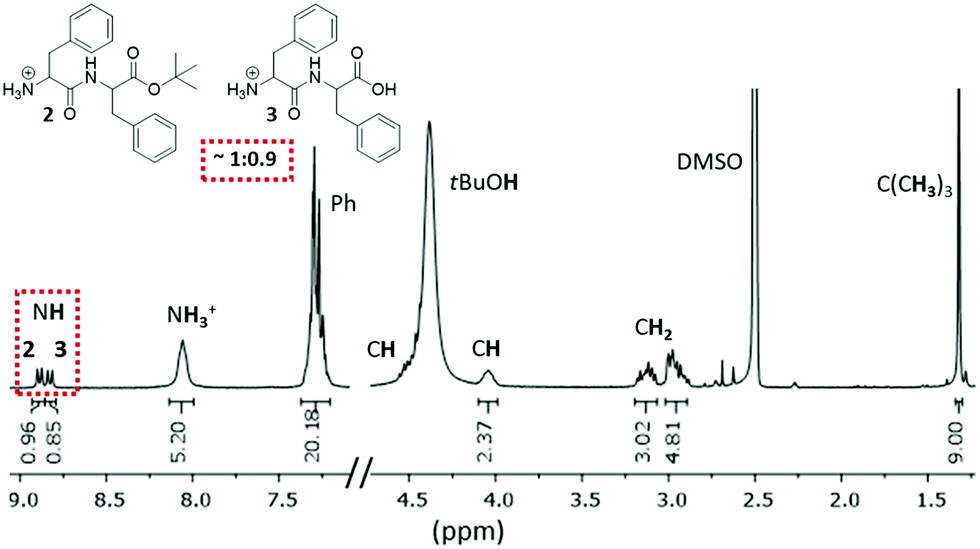 | ||
| Fig. 2 1H NMR (300 MHz, d6-DMSO) spectrum of xerogel I. Gelation is induced by products 2 and 3. | ||
To assess potential changes in the ratio of compounds 2 and 3 and their impact on the gel's lifetime, we recorded the NMR spectra of dissolved dried xerogels, prepared at different time points, ranging from the formation of the gel (day 1) until the transition to the solution phase (day 4). Although both compounds 2 and 3 were present in the system, their ratio appeared to change randomly over 4 days (Fig. 3). Interestingly, the dynamic interconversion between dipeptides 2 and 3 does not seem to be responsible for the collapse of the gel but rather the formation of tBuOH over time.
 | ||
| Fig. 3 1H NMR (300 MHz, d6-DMSO) spectra of xerogels over 4 days. | ||
Indeed, when we added tBuOH on the surface of freshly prepared organogels, they collapsed within 48 h, in contrast to the untreated material. Additional control experiments with water showed that the gel collapses almost immediately upon addition. The water in the system originates from the acid solution and solvents. However, its amount is insufficient to break the gel structure as the untreated organogel collapses after 4 days. Upon addition of the acid, part of the formed tert butyl cation (tBu+) re-esterifies Boc–Phe–Phe 4 towards Boc–Phe–Phe–OtBu 1 (Fig. 1) in a rapid process, as per the given mechanism. Based on the control experiments and the obtained NMR spectra, we, therefore, suggest that the formation of tBuOH at a slower pace is responsible for the re-esterification of Phe–Phe 3 to the mono-protected ester Phe–Phe–OtBu 2.
To clarify the observed random evolution of compounds 2 and 3 over time and their contribution to gel formation, we tested whether components 2 and 3 could form alone a self-supporting gel in tBuOAc. In the absence of sulfuric acid, ester 2 did not gel, indicating that compound 2 cannot form gels independently. However, when 1.0 equivalent of the acid was added, compound 3 formed in situ, leading to a mixture of 2 and 3 at a 0.9:1 ratio that once again yielded a self-supporting organogel. NMR analysis of the corresponding xerogel (II) showed qualitative spectra similar to xerogel (I) (Fig. S5 and S6, ESI†). In contrast, Phe–Phe 3 did not gel in tBuOAc even after the addition of the acid. Thus, to induce gelation under the given conditions, the presence of both compounds 2 and 3 is required.
Gelation is dynamically driven by the presence of acid in the system. The organogel appears to be thermodynamically meta-stable since supramolecular forces between derivatives 2 and 3 develop relatively rapidly (12 h), leading to gelation. The observed interconversion between gelators 2 and 3 does not affect the stability of the gel. In contrast, the formation of tBuOH, which occurs more slowly over four days, converts the gel to a solution, as it seems to act antagonistically against tBuOAc (Fig. 4). Therefore, the presence of tBuOAc, which reverses the conversion of 2 into 3, the process that otherwise forms tBuOH, effectively acts as a brake on the process. In principle, the balance between the “accelerating” sulfuric acid and the “braking” tBuOAc will dictate the gel's metastability. This means that the degradation time could be controlled, for example, by changing the acid concentration. Indeed, when only 0.5 equivalents of acid were used, the lifetime of the gels extended beyond 4 weeks (Table S2, ESI†). Such an approach might be beneficial for applications that require the formation and controlled degradation of soft materials at a given time.24–27
 | ||
| Fig. 4 Transient assembly: precursor 1 is activated by acid “accelerator” towards gelators 2 and 3. The solvent (tBuOAc) acts as a “brake”, reversing the conversion of 2 into 3 and preventing the formation of tBuOH, which otherwise causes the gel to decay. | ||
According to the literature, diphenylalanine-based nanostructures adopt β-sheet conformations when assembled, as per the reported positions of the amide I bands.28–30 Here, the xerogel shows a peak at 1661 cm−1 suggesting the potential formation of 310-helices31 rather than a parallel or antiparallel β-sheet secondary structure.
In addition, we performed swelling studies on the organogel in tBuOAc to assess its stability. Two gel specimens at concentrations of 0.05 M (MGC) and 0.1 M were used. The calculated swelling degree (SD-%) of each sample against time revealed that swelling is concentration-dependent (Fig. S4, ESI†). At the MGC (0.05 M), swelling reached a plateau after sixteen days, whereas at a higher concentration (0.1 M), no swelling was observed, even after ten days of continuous measurements. The swollen material absorbed solvent up to 6% of its initial weight, demonstrating a high degree of elasticity. Rheology studies at the MGC before swelling revealed a storage modulus (G′) of 10890 Pa and a loss modulus (G′′) of 2474 Pa, respectively (Fig. S12, ESI†).
Although the standard organogel has a lifespan of four days, both materials remained intact for twenty days during the swelling studies. Seven months later, the swollen gel was still stable. The lifespan expansion suggests that the solvent treatment affected the consistency and potentially the materials' supramolecular network. Therefore, we recorded the NMR spectra of the swollen gel (Fig. S8 and S9, ESI†) to assess potential changes in its consistency. In contrast to the initial organogel, only ester 2 was detected. It seems likely that adding extra solvent and the material's swelling helps prevent the deprotection of compound 2 towards 3. Indeed, removing excess solvent from the gel's surface during each measurement will also decrease the acid concentration, effectively removing some of the “accelerator” from the system. Meanwhile, tBuOAc forms the tertiary cation in excess and regenerates ester 2, acting as a “brake”. We reason that these processes combine to extend the lifetime of the metastable gel. Interestingly, when the mixture of 2 and 3 is treated with tBuOAc in this way, compound 2 can maintain the gel behaviour, whereas, on its own, it cannot establish a gel. This indicates, as is often seen for metastable gels that the assembly pathway plays an important role in controlling the material's performance.32
Notably, during the swelling studies, the conditions changed since sulfuric acid concentration was now present at less than 1.0 equivalent. To relate the results of swelling studies with potential alterations of the gel's network, we performed scanning electron microscopy (SEM) imaging of the gel samples (Fig. 5). Images taken immediately after the gel formation revealed a dense three-dimensional network of branched, entangled, plated ribbons. The swollen material after twenty days of treatment showed elongated, fine fibres in coil-coiled constructions. Seven months later, the network consisted of thicker fibres intertwined into wider bundles. This suggests the slow dynamic evolution of the structure over time.33
 | ||
| Fig. 5 SEM imaging after (A) 1 day; (B) 20 days; (C) 7 months. Inset depicts the corresponding bulk gels. The double layer (B and C) is a mirror effect due to the camera's angle. | ||
Our studies prove a gelation system, which evolves dynamically over time through a set of coupled reactions, some of which are in equilibrium. Sulfuric acid is the reaction “accelerator”, driving the deprotection reactions of NHBoc and tBu-ester forward. The solvent (tBuOAc) acts as a “brake” by effectively reversing the ester deprotection. This prevents the formation of tBuOH, which otherwise appears to trigger the gel-to-sol transition. The organogel is thermodynamically meta-stable and responds to the presence of the “accelerator” or the “brake”. For example, swelling of the material with additional solvent limits the hydrolysis of the ester and the formation of tBuOH, thus, extending its lifespan and altering its supramolecular network. Therefore, sample-handling procedures could introduce, to some extent, tuneability to the micro- and macroscopic properties of gels. This system constitutes a rare example in which the solvent does not only play a passive role in supporting the gelation process but is intimately involved in the chemical equilibria, which underpin the dynamic gelation event itself and helps trigger gel assembly/breakdown.
The authors would like to acknowledge Jane and Aatos Erkko Foundation for supporting the current work.
E. S. and R. C. designed and performed the synthesis, characterization and gelation experiments with J. S. and wrote the original draft, C. C. P. did the rheological measurements and data interpretation with E. S., E. K. did the mass spectrometric studies and analysis, A. J. did the microscopic imaging and visualization, D. S. is responsible for rheological resources, data interpretation and review and editing of the manuscript, M. P. supervised the work of J. S. and acquired the funding and resources, M. N. supervised the work of R. C., was responsible for project management and review and editing of the manuscript with input from all authors.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Boekhoven, W. E. Hendriksen, G. J. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349(6252), 1075–1079 CrossRef CAS PubMed.
- C. A. Angulo-Pachón and J. F. Miravet, Chem. Commun., 2016, 52, 5398–5401 RSC.
- S. Dhiman and S. J. George, Bull. Chem. Soc. Jpn., 2018, 91, 687–699 CrossRef CAS.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., 2010, 122, 4935–4938 CrossRef.
- G. Ragazzon and L. J. Prins, Nat. Nanotechnol., 2018, 13, 882–889 CrossRef CAS PubMed.
- W. A. Ogden and Z. Guan, ChemSystemsChem, 2020, 2, e1900030 CrossRef CAS.
- B. Rieß, R. K. Grötsch and J. Boekhoven, J. Chem., 2020, 6, 552–578 Search PubMed.
- F. della Sala, S. Neri, S. Maiti, J. L. Y. Chen and L. J. Prins, Curr. Opin. Biotechnol., 2017, 46, 27–33 CrossRef CAS PubMed.
- T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54(45), 13258–13262 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- T. Muraoka, C. Y. Koh, H. Cui and S. I. Stupp, Angew. Chem., Int. Ed., 2009, 48, 5946–5949 CrossRef CAS PubMed.
- S. Chen, R. Costil, F. K. C. Leung and B. L. Feringa, Angew. Chem., Int. Ed., 2021, 60, 11604–11627 CrossRef CAS PubMed.
- T. Kar, S. K. Mandal and P. K. Das, Chem. – Eur. J., 2011, 17, 14952–14961 CrossRef CAS PubMed.
- G. Aykent, C. Zeytun, A. Marion and S. Özçubukçu, Sci. Rep., 2019, 9, 1–8 Search PubMed.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS PubMed.
- V. Nele, C. E. Schutt, J. P. Wojciechowski, W. Kit-Anan, J. J. Doutch, J. P. K. Armstrong and M. M. Stevens, Adv. Mater., 2020, 32, 1905914 CrossRef CAS PubMed.
- S. Panja, K. Boháčová, B. Dietrich and D. J. Adams, Nanoscale, 2020, 12, 12840–12848 RSC.
- N. Singh, B. Lainer, G. J. M. Formon, S. De Piccoli and T. M. Hermans, J. Am. Chem. Soc., 2020, 142(9), 4083–4087 CrossRef CAS PubMed.
- D. Spitzer, L. L. Rodrigues, D. Straßburger, M. Mezger and P. Besenius, Angew. Chem., Int. Ed., 2017, 56(48), 15461–15465 CrossRef CAS PubMed.
- T. Das, M. Häring, D. Haldar and D. Díaz Díaz, Biomater. Sci., 2018, 6, 38–59 RSC.
- S. Marchesan, A. V. Vargiu and K. E. Styan, Molecules, 2015, 20, 19775–19788 CrossRef CAS PubMed.
- L. S. Lin, T. L. Jr, E. E. De Laszlo, Q. Truong, T. Kamenecka and W. K. Hagmann, Tetrahedron Lett., 2000, 41, 7013–7016 CrossRef CAS.
- B. Rieß and J. Boekhoven, ChemNanoMat, 2018, 4, 710–719 CrossRef.
- M. Tena-Solsona, B. Rieß, R. K. Grötsch, F. C. Löhrer, C. Wanzke, B. Käsdorf, A. R. Bausch, P. Müller-Buschbaum, O. Lieleg and J. Boekhoven, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- J. L. Y. Chen, S. Maiti, I. Fortunati, C. Ferrante and L. J. Prins, Chem. – Eur. J., 2017, 23, 11549–11559 CrossRef CAS PubMed.
- T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Isr. J. Chem., 2005, 45, 363–371 CrossRef CAS.
- X. Yan, P. Zhu and J. Li, Self-assembly and application of diphenyl-alanine-based nanostructures, Chem. Soc. Rev., 2010, 39, 1877–1890 RSC.
- R. Huang, R. Su, W. Qi, J. Zhao and Z. He, Nanotechnology, 2011, 22, 245609 CrossRef PubMed.
- J. Kong and S. Yu, Acta Biochim. Biophys. Sin., 2007, 39, 549–559 CrossRef CAS PubMed.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. La Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS PubMed.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, synthetic protocols, NMR, FT-IR and HR-MS spectra, gelation trials, swelling and rheology plots. See DOI: 10.1039/d1cc04021a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |