
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step methylation of aromatic phosphorus heterocycles: synthesis and crystallographic characterization of a 1-methyl-phosphininium salt†‡
Lukas
Fischer§
,
Friedrich
Wossidlo§
,
Daniel
Frost
,
Nathan T.
Coles
 ,
Simon
Steinhauer
,
Sebastian
Riedel
* and
Christian
Müller
*
,
Simon
Steinhauer
,
Sebastian
Riedel
* and
Christian
Müller
*
Institut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstrasse 34/36, Berlin 14195, Germany. E-mail: s.riedel@fu-berlin.de; c.mueller@fu-berlin.de
First published on 6th August 2021
Abstract
For the first time, the direct synthesis of 1-methyl-phosphininium salts has been achieved by reacting aromatic λ3,σ2-phosphinines with the readily available dimethyl chloronium salt [(CH3)2Cl]+[Al(OTeF5)4]−. The remarkably high electrophilicity of the alkylation reagent in combination with the weakly coordinating pentafluoro-orthotelluratoaluminate anion offers excellent conditions for this one-step approach. Our simple and quantitative access to 1-methyl-phosphininium salts will pave the way to explore the chemistry of such reactive species in more detail.
Tertiary phosphines (PR3) react readily with alkyl halides R-X or Meerwein reagents [R3O]+[BF4]− to form the corresponding quaternary alkyl-phosphonium salts [R3P-R]+[X/BF4]−.1 These compounds are thermodynamically and kinetically stable solids, that are soluble in polar solvents and provide a range of applications. For instance, they are widely used to generate phosphonium ylides for Wittig reactions, as ionic liquids, phase-transfer catalysts, and phosphorus-based flame retardants, or for the stabilization of highly reactive anions.1 Arylphosphonium cations [Ar3P-R]+ have lipophilic properties and the corresponding salts find biological applications.2 The facile conversion of tertiary phosphines with appropriate alkyl halides to phosphonium salts is typically related to the pronounced basicity and nucleophilicity of phosphines in the SN2-type alkylation reaction. These properties are generally strongest in trialkylphosphines and decrease with aryl-substitution at the phosphorus atom.
In contrast to classical phosphines, λ3,σ2-phosphinines (phosphabenzenes), containing a phosphorus atom in low coordination, are extremely weak bases and very poor nucleophiles.3 This can be attributed to the rather high 3s-character of the phosphorus lone pair (64% 3s character in C5H5P versus 29% 2s character in pyridine C5H5N).4 Reactions of electrophiles at the phosphorus atom are therefore challenging and it is very difficult to access phosphininium ions directly, which are assumed to occur as intermediates in the formation of λ5-phosphinines.5 Consequently, little is still known about phosphininium cations. Even though 2,4,6-tris-tBu-λ3-phosphinine can be protonated at the phosphorus atom with the in situ generated, non-oxidizing superacid H[CHB11Me5Cl6] to afford the 1H-phosphininium salt [H(C5H2tBu3P)]+ [CHB11Me5Cl6]− (1-H, Chart 1), 1-phosphinines cannot be directly alkylated to 1R-phosphininium cations with classical reagents, such as alkyl halides or Meerwein salts.6,7
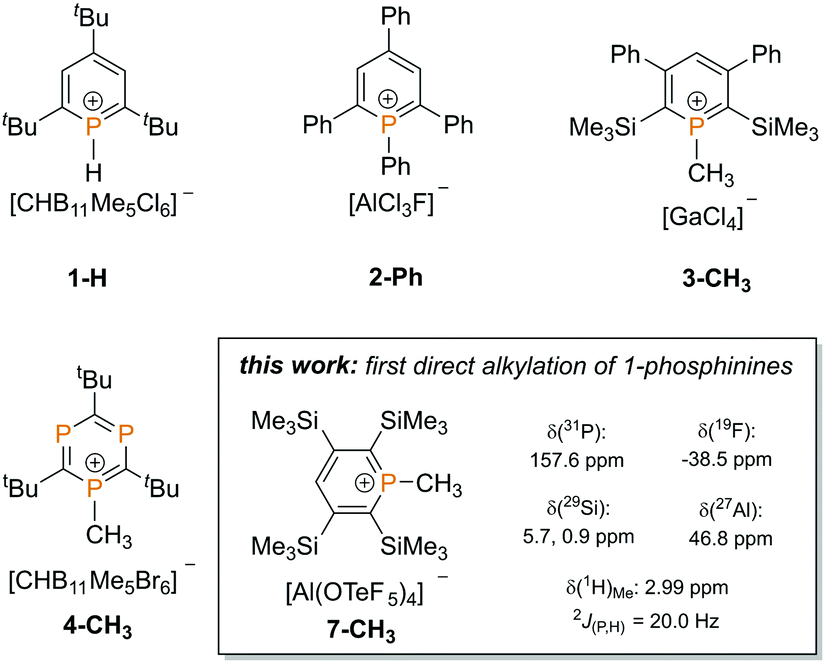 | ||
| Chart 1 Known phosphininium salts and a brief overview of this work. | ||
The first synthesis of a 1R-phosphininium salt was carried out starting from 1-fluoro-1-phenyl-2,4,6-triphenylphosphinine.8 Dimroth et al. showed that the fluoride anion can be abstracted with aluminium trichloride, leading to the formation of 2-Ph (Chart 1). The complete characterization failed due to the low solubility and high reactivity of the 1-phenyl-2,4,6-triphenylphosphininium ion. In 2003, Le Floch and co-worker were able to obtain 1-methylphosphinium ions, such as 3-CH3via an adapted route and could fully characterize the desired compounds.9 The only direct alkylation of a phosphinine derivative was reported by Reed et al. in 2008. Reaction of a more electron rich triphosphinine with the carborane-based strong electrophilic methylating agent Me[CHB11Me5Br6] led to compound 4-CH3, which was also characterized crystallographically (Chart 1).6
We recently found that the basicity and nucleophilicity of phosphinines can be increased significantly by introducing Me3Si-substituents to the aromatic phosphorus heterocycle.10 Due to the strongly electron donating character and the related β-silyl-effect of this group, we were able to access phosphinine selenides and could also fully characterize phosphinine–borane adducts for the first time.11 This prompted us to investigate the reaction between a series of phosphinines with the strongly electrophilic methylation reagent [(CH3)2Cl]+[Al(OTeF5)4]−, containing the weakly coordinating and chemically inert pentafluoroortho-telluratoaluminate anion, which should be ideally suited to isolate the rather reactive 1-methyl-phosphininium cation. This dimethyl chloronium salt, recently reported by one of the authors, has already been proven to efficiently alkylate the weakly nucleophilic phosphines P(CF3)3 and PF3.12 A simple and direct access to 1-alkyl-phosphininium cations would make detailed investigations on their properties and reactivity more feasible. We report here the first example of direct alkylation for a series of 1-phosphinines with [(CH3)2Cl]+[Al(OTeF5)4]−. All 1-methyl-phosphininium salts were investigated spectroscopically and the most stable salt studied could be characterized crystallographically.
The strongest accessible methylating agent in the series of dimethyl halonium ions is the dimethyl chloronium cation. It can be obtained by reacting chloromethane (CH3Cl) with a Brønsted superacid. This method has already been used with the readily available teflate-based superacid [H][Al(OTeF5)4]solv., which is generated in situ from AlEt3 and HOTeF5 in a suitable solvent.13 The neat [(CH3)2Cl]+[Al(OTeF5)4]− salt is available in gram quantities and, importantly, it is a non-oxidizing reagent.
We decided to investigate three differently substituted phosphinines (5–7) in their reactivity towards the dimethyl chloronium salt. We were particularly interested in whether the increased basicity and nucleophilicity of the phosphorus atom from compound 5 → 7 is crucial for the alkylation reaction (Fig. 1).
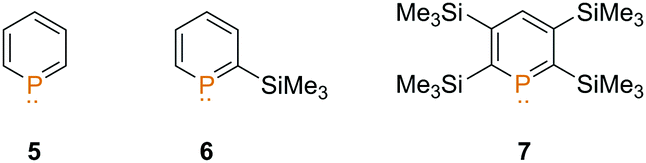 | ||
| Fig. 1 Phosphinines 5–7 used in the methylation reaction. | ||
As recently demonstrated by us, the parent compound C5H5P (5) is the least basic phosphinine in this series and exhibits the lowest gas phase basicity (GB = −ΔG0 of protonation a T = 298.15 K, Table 1).11 With a growing number of Me3Si-substituents at the aromatic phosphorus heterocycle, the gas phase basicity significantly increases. Compound 7 shows the highest calculated gas phase basicity among the phosphinines and is even more basic than pyridine in the gas phase (898.5 kJ mol−1).14 In addition, the increased gas phase basicity of Me3Si-substituted phosphinines is concomitant with an energetic destabilization of the P-lone pair (HOMO−1). This should particularly render phosphinines 6 and 7 as ideal candidates for methylation reactions.
| Compound | Lone pair energya [eV] | Gas-phase basicityb [kJ mol−1] | CH3+ affinityc [kJ mol−1] |
|---|---|---|---|
| a B3LYP/def2-TZVPP level of theory calculated with solvent model COSMO (εR = 1.84). b DLPNO-CCSD(T)-MP2 compound method. c B3LYP/def2-TZVPP level of theory calculated in the gasphase. | |||
| 5 | −7.40 | 791.1 | 458.0 |
| 6 | −7.20 | 831.9 | 489.0 |
| 7 | −6.54 | 905.5 | 513.0 |
| Pyridine | −7.13 | 898.5 | 501.7 |
| PPh3 | −5.91 | n.d. | 597.8 |
According to quantum-chemical calculations on the B3LYP/def2-TZVPP level of theory, the methyl cation affinity of phosphinines 5–7 is further consistent with the calculated gas phase basicities and the respective lone pair energies (Table 1, eqn (1); LB = Lewis Base).
| LB + CH3+ → LB-CH3+ | (1) |
As a matter of fact, the unsubstituted phosphinine 5 has the lowest methyl cation affinity in this series of phosphinines, while the introduction of one silyl substituent already increases this value by 31 kJ mol−1 (Table 1). The methyl cation affinity of phosphinine 7 exceeds even that of pyridine, which can already be methylated by a variety of conventional alkylating agents. As expected, the methyl cation affinities of phosphinines 5–7 are significantly lower compared to triphenylphosphine (PPh3).
Phosphinines 5–7 were synthesized according to literature procedures.10,15 Single crystals of 7, suitable for X-ray diffraction, could be obtained from a concentrated solution of this compound in acetonitrile. The solid state structure of 7 (Fig. S18, ESI‡) reveals that the aromatic phosphorus heterocycle is fully planar, despite the fact that a total of four sterically rather demanding Me3Si-groups are present. The methyl-substituents of the Me3Si-groups are clearly staggered. The values found for the P–C and C–C bond distances as well as for the internal C(1)–P(1)–C(5) angle are similar to the ones reported for other phosphinines.16
The choice of solvent for the methylation of phosphinines with [(CH3)2Cl]+[Al(OTeF5)4]− is limited, as the high electrophilicity of the methylating agent allows not only the methylation of weak Lewis bases, but also the reaction with conventional solvents. Thus, dichloromethane, acetonitrile, ether and aromatic solvents (e.g. benzene, toluene) are not suitable. Solvents such as sulfur dioxide, sulphuryl chloride fluoride, chloromethane and saturated hydrocarbons, on the other hand, are rather inert towards the dimethyl chloronium salt. For our studies, it turned out that a mixture of chloromethane and n-pentane is an ideal solvent system for our purpose.
The dimethyl chloronium salt was first dissolved in chloromethane, while a solution of the phosphinine in n-pentane was prepared and cooled down to T = −110 °C. Subsequently, the pentane solution was added to the solution of the dimethyl chloronium salt in chloromethane via a PFA (perfluoroalkoxy) tube. The mixture was stirred at low temperatures until it turned from yellow to orange, and analyzed by means of NMR spectroscopy as well as mass spectrometry (TOF-MS-ES+).
Much to our surprise, the parent phosphinine 5 already reacts at T = −80 °C cleanly and quantitatively with the dimethyl chloronium salt to the corresponding 1-methyl phosphininium salt 5-CH3 (Scheme 1).
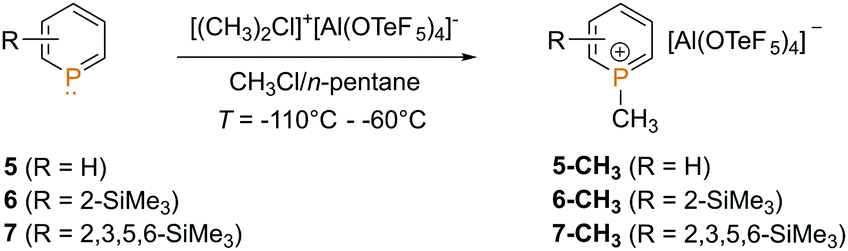 | ||
| Scheme 1 Reaction of phosphinines 5–7 with [(CH3)2Cl][Al(OTeF5)4]. | ||
Fig. 2 shows the 31P NMR spectrum of 5 in comparison to 5-CH3. The direct methylation of the phosphorus atom is accompanied by a strong upfield shift of the phosphorus signal from δ(ppm) = 206.4 (5) to δ(ppm) = 136.6 (5-CH3; δΔ = 70 ppm). Apparently, the dimethyl chloronium salt is such a strong alkylating reagent, that even 5, which shows a very low basicity and nucleophilicity, can be methylated efficiently.
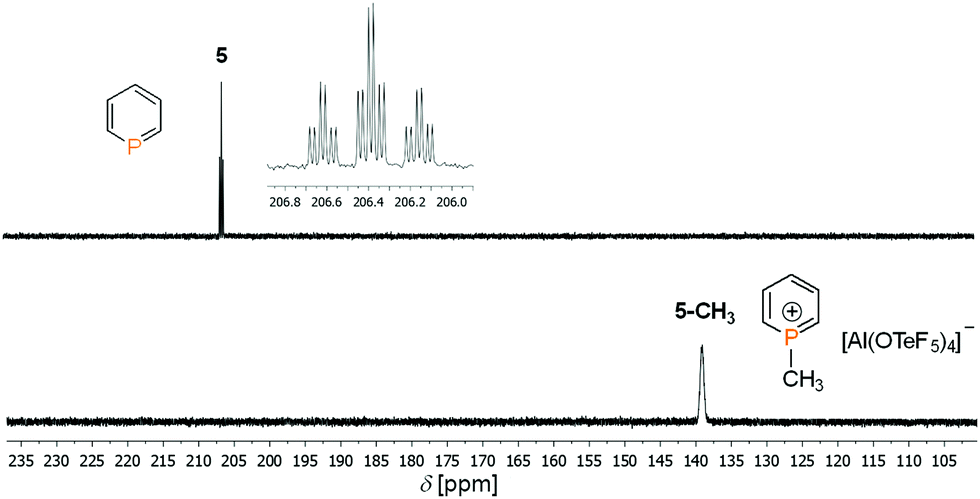 | ||
| Fig. 2 31P NMR of 5 (25 °C) and 5-CH3 (T = −70 °C) in CD2Cl2. | ||
However, phosphininium salt 5-CH3 starts to decompose in solution to unidentified products above T = −70 °C and could only be characterized by low-temperature NMR spectroscopy (Table S1, ESI‡).
In the 1H NMR spectrum of 5-CH3, the signals of the protons of the heterocycle are found in the aromatic region between δ(ppm) = 8.0–8.7 and partially overlap. The signal of the methyl group is found at δ(ppm) = 3.16 and splits into a doublet with a coupling constant of 2J(P,H) = 24.0 Hz. The chemical shift and the coupling constant is in agreement with the values found for 3-CH3 (Fig. 1). In the 13C{1H} NMR spectrum, the doublet at δ(ppm) = 7.18 can be assigned to the methyl group at the phosphorus atom. The three doublets for the carbon atoms of the heterocycle can be detected between δ(ppm) = 125–145. Their chemical shifts indicate that the phosphinine preserves its aromaticity even after methylation. This subject has already been addressed by Le Floch and co-worker for 3-CH3.9
Reaction of the Me3Si-substituted phosphinines 6 and 7 with [(CH3)2Cl]+[Al(OTeF5)4]− were more interesting, as the products 6-CH3 and 7-CH3 seem to be both kinetically and thermodynamically more stable. Interestingly, 7-CH3 can be handled at room temperature for approximately 5 hours without noticeable decomposition, with full decomposition occurring after 4 days, which is in contrast to 5-CH3. We attribute these properties to the presence of Me3Si-groups at the phosphorus heterocycle, which provide steric protection of the reactive P-CH3+ moiety. Additionally, the Me3Si-substituents can contribute to the thermodynamic stabilization of the phosphininium cation due to the electron donating nature of the Me3Si-groups, making its access much more feasible. The methylation of 6 and 7 again proceeds cleanly and quantitatively at low temperature. Decomposition of all three phosphininium salts occurs unselectively and gives mixtures of unidentified compounds in addition to some black, insoluble precipitate. 6-CH3 and 7-CH3 were fully characterized by means of NMR spectroscopy and the data are similar to what was observed for 5-CH3 (Table S1, ESI‡). Finally, we characterized all three phosphininium salts also by means of high resolution mass spectrometry, which confirmed the formation of the phosphininium cations (Table S1, ESI‡).
Single crystals of 7-CH3 were obtained (monoclinic space group Cc) could be obtained from a solution of 7-CH3 in dichloromethane at T = −80 °C. The result of the X-ray crystal structure determination is depicted in Fig. 3 and unequivocally confirms the direct, clean and quantitative methylation of a λ3-phosphinine with the dimethyl chloronium salt [(CH3)2Cl]+[Al(OTeF5)4]−.
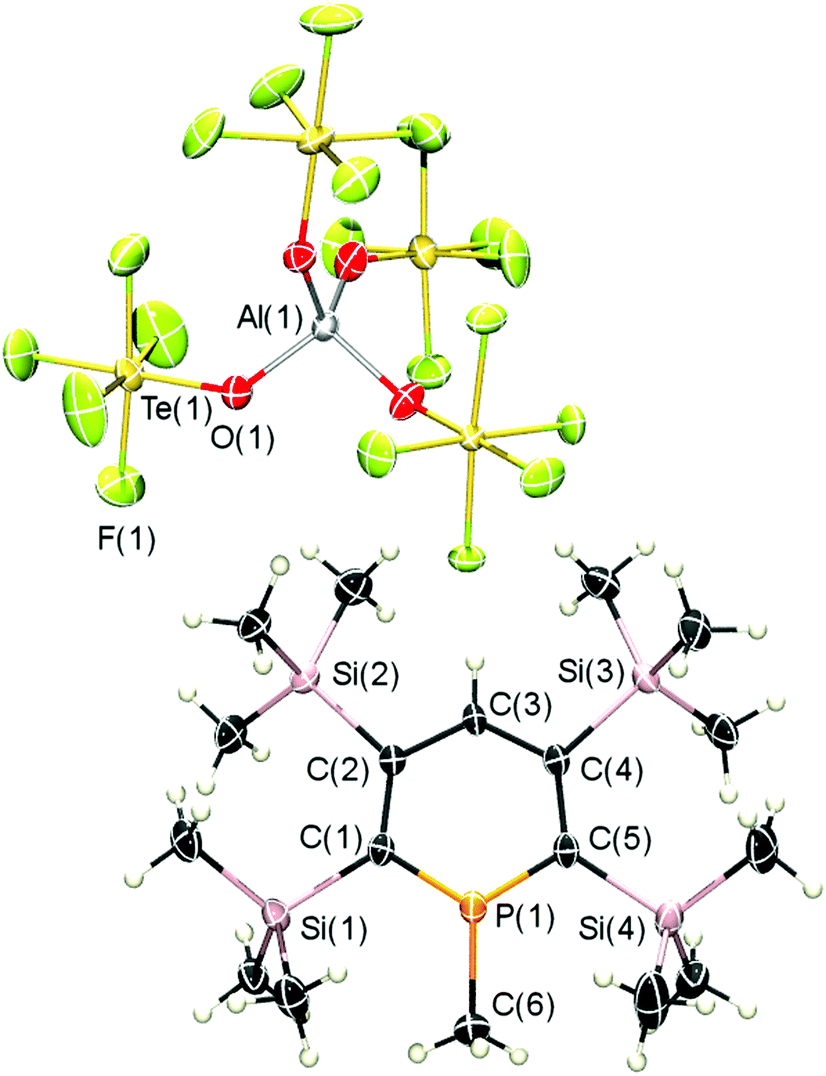 | ||
| Fig. 3 Molecular structure of 7-CH3 in the crystal. Displacement ellipsoids are shown at the 50% probability level. Selected bond lengths (Å) and angles (°): P(1)–C(6): 1.807(4); P(1)–C(1): 1.696(3); P(1)–C(5): 1.693(3); C(1)–C(2): 1.419(4); C(2)–C(3): 1.440(4); C(3)–C(4): 1.454(4); C(4)–C(5): 1.414(4). C(1)–P(1)–C(5): 118.20(16); C(2)–C(3)–C(4): 127.2(3). | ||
Interestingly, C(6) deviates 11.75° from the plane of the heterocyclic ring (Fig. S19, ESI‡). Moreover, the bond lengths and angles of the phosphinine ring differ from those found for phosphinine 7 (Fig. S18, ESI‡). As a matter of fact, the phosphorus–carbon bonds are significantly shorter in the phosphininium cation than in the corresponding phosphinine (1.696(3) and 1.693(3) Å vs. 1.745(11) and 1.707(11) Å). In addition, the C(1)–P(1)–C(5) angle is wider in the cation of 7-CH3 than in 7 (118.20(16)° vs. 106.0(4)°). Similar findings were already observed for 3-CH3 and the corresponding phosphinine and explained with a stronger formal sp2 hybridization of the phosphorus atom in the phosphininium cation in comparison to the neutral λ3-phosphinine.9 With respect to the anion, the aluminium atom shows a distorted tetrahedral geometry and is surrounded by the four OTeF5-groups with O–Al–O bond angles of 106.4(5) to 110.6(1)°.
We anticipate, that the strong deviation of C(6) from the planar phosphorus heterocycle (11.75°) is due to the presence of the four Me3Si-groups. As already observed for phosphinine 7 (Fig. S18, ESI‡), the Me3Si-groups are also staggered in the cation of 7-CH3 (Fig. 3). Consequently, the methyl groups of the two ortho-Me3Si-substituents push the CH3-substituent at the phosphorus atom below the plane of the heterocycle, as can be seen from the side view of the cation in Fig. S19 (ESI‡).
In order to clarify this observation, we performed structure optimizations of both the phosphininium cation of 7-CH3 and of 5-CH3, containing the parent ion [C5H5P–CH3]+ (Fig. S20, ESI‡). The DFT-calculations on the cation of 7-CH3 reproduce the crystallographic data very well. The deviation of C(6) from the heterocyclic ring is slightly larger in the calculated structure than in the crystal data (17.09° vs. 11.75°). In contrast, almost no bending of C(6) can be observed in the cation of 5-CH3 (Fig. S20b, ESI‡). We can therefore conclude that the observed geometrical features in the phosphininium cation of 7-CH3 are due to steric effects, caused by the four sterically demanding Me3Si-substituents.
A comparison of the Kohn–Sham-LUMOs of C5H5P–CH3+ and C5H5N–CH3+ reveals an interesting difference between these species (Fig. 4). One of the three C–H-groups of the methyl-substituent in C5H5P–CH3+ seems to participate in hyperconjugative effects with the formally positively charged phosphorus atom, which might cause an additional stabilization of the P–C bond (Fig. 4a). This interaction is much less pronounced in the pyridininium cation C5H5N–CH3+ (Fig. 4b).
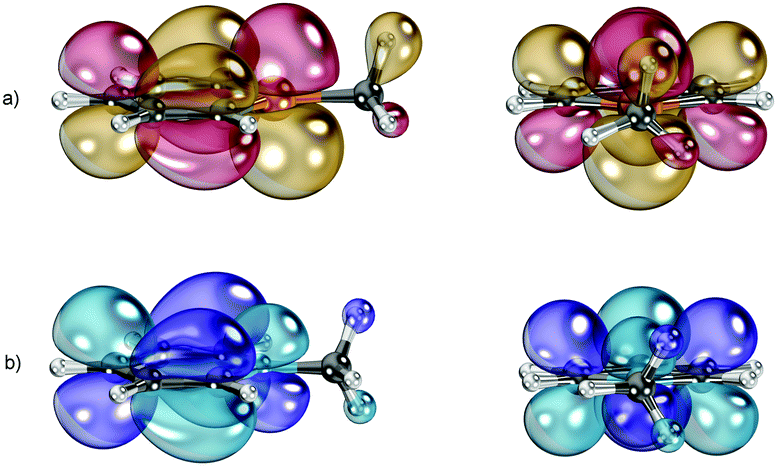 | ||
| Fig. 4 LUMO of C5H5P–CH3+ (a) and C5H5N–CH3+ (b). Side view (left) and front view (right). B3LYP/def2-TZVP level of theory. | ||
In conclusion, we could demonstrate for the first time, that 1-phosphinines can be directly methylated using the stable and easily synthesized dimethyl chloronium salt [(CH3)2Cl]+[Al(OTeF5)4]−, which acts as a very strong electrophile. All new compounds could be characterized by means of NMR spectroscopy and mass spectrometry. In addition, one of the novel 1-methyl-phosphininium salts could be characterized crystallographically. The results of our reactivity studies confirm the hypothesis that Me3Si-substituents at the aromatic phosphorus heterocycle have a clear influence on the kinetic and thermodynamic stability of the corresponding products, in addition to their nucleophilicity.11
The remarkably high electrophilicity of the dimethyl chloronium cation as well as the high kinetic and thermodynamic stability of the weakly coordinating pentafluoro-orthotelluratoaluminate offer excellent properties for the one-step synthesis of stable 1-methyl phosphininium salts. This is an important observation, as λ3-phosphinines normally suffer from an extremely low basicity and nucleophilicity, making their reactivity towards electrophiles very challenging. Our results on a facile synthetic approach to 1-methyl-phosphininium cations can lead to a deeper understanding of the properties and reactivities of such species as the simple and quantitative access to kinetically stable phosphininium salts is now feasible. Additionally, our results may pave the way to apply the dimethyl chloronium salt [(CH3)2Cl]+[Al(OTeF5)4]− for the direct methylation of other low coordinate phosphorus donors of low basicity and nucleophilicity.
Conflicts of interest
There are no conflicts to declare.Notes and references
- V. Iaroshenko, Organophosphorus Chemistry, Wiley-VCH, Weinheim, 2019 Search PubMed.
- M. Lukáč, M. Pisárčik, M. Garajová, M. Mrva, A. Dušeková, A. Vrták, R. Horáková, B. Horváth and F. Devínsky, J. Surfactants Deterg., 2020, 23, 1025 CrossRef.
- (a) J. Waluk, H.-P. Klein, A. J. Ashe and J. Michl, Organometallics, 1989, 8, 2804 CrossRef CAS; (b) C. Batich, E. Heilbronner, V. Hornung, A. J. Ashe, D. T. Clark, U. T. Cobley, D. Kilcast and I. Scanlan, J. Am. Chem. Soc., 1973, 99, 928 CrossRef.
- P. Le Floch, Coord. Chem. Rev., 2006, 250, 627 CrossRef CAS.
- G. Märkl, K. Hock and D. Matthes, Chem. Ber., 1983, 116, 445 CrossRef.
- Y. Zhang, F. S. Tham, J. F. Nixon, C. Taylor, J. C. Green and C. A. Reed, Angew. Chem., Int. Ed., 2008, 47, 3801 CrossRef CAS PubMed.
- G. Märkl, F. Lieb and A. Merz, Angew. Chem., Int. Ed. Engl., 1967, 6, 87 CrossRef.
- T. N. Dave, H. Kaletsch and K. Dimroth, Angew. Chem., 1984, 96, 984 CrossRef CAS.
- A. Moores, L. Ricard and P. Le Floch, Angew. Chem., Int. Ed., 2003, 42, 4940 CrossRef CAS.
- (a) M. H. Habicht, F. Wossidlo, M. Weber and C. Müller, Chem. – Eur. J., 2016, 22, 12877 CrossRef CAS; (b) M. H. Habicht, F. Wossidlo, T. Bens, E. A. Pidko and C. Müller, Chem. – Eur. J., 2018, 24, 944 CrossRef CAS.
- F. Wossidlo, D. S. Frost, J. Lin, N. T. Coles, K. Klimov, M. Weber, T. Böttcher and C. Müller, Chem. – Eur. J., 2021 DOI:10.1002/chem.202102390.
- S. Hämmerling, G. Thiele, S. Steinhauer, H. Beckers, C. Müller and S. Riedel, Angew. Chem., Int. Ed., 2019, 58, 9807 CrossRef PubMed.
- A. Wiesner, T. W. Gries, S. Steinhauer, H. Beckers and S. Riedel, Angew. Chem., Int. Ed., 2017, 56, 8263 CrossRef CAS PubMed.
- (a) E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413 CrossRef CAS; (b) J. Schröder, D. Himmel and T. Böttcher, Chem. – Eur. J., 2017, 23, 10763 CrossRef.
- N. Avavari, P. Le Floch and F. Mathey, J. Am. Chem. Soc., 1996, 118, 11978 CrossRef.
- See for example: C. Müller, L. E. E. Broeckx, I. de Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 187 CrossRef.
Footnotes |
| † In memoriam of Prof. Dr. Manfred Regitz. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2062243 and 2062244. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc03892c |
| § These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |