
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCavity-promotion by pillar[5]arenes expedites organic photoredox-catalysed reductive dehalogenations†
Maximilian
Schmidt
 a and
Birgit
Esser
*abc
a and
Birgit
Esser
*abc
aInstitute for Organic Chemistry, University of Freiburg, Albertstraße 21, 79104 Freiburg, Germany. E-mail: besser@oc.uni-freiburg.de; www.esser-lab.uni-freiburg.de
bFreiburg Materials Research Center, University of Freiburg, Stefan-Meier-Str. 21, 79104 Freiburg, Germany
cFreiburg Center for Interactive Materials and Bioinspired Technologies, University of Freiburg, Georges-Köhler-Allee 105, 79110 Freiburg, Germany
First published on 12th August 2021
Abstract
The efficiency of the photo-induced electron transfer in photoredox catalysis is limited by the diffusional collision of the excited catalyst and the substrate. We herein present cavity-bound photoredox catalysts, which preassociate the substrates, leading to significantly shortened reaction times. A pillar[5]arene serves as the cavity and phenothiazine as a catalyst in the reductive dehalogenation of aliphatic bromides as a proof of concept reaction.
Photoredox catalysts (PRCs) allow the conversion of light into chemical energy.1,2 They are considered a more sustainable alternative to traditional catalyst systems, using light as a non-hazardous and environmentally friendly energy source instead of high temperatures or harsh reaction conditions. Significant advances have been achieved in recent years using PRCs in synthetic organic chemistry.3–5 Both transition metal complexes6,7 as well as organic dyes8–10 have successfully been employed in a variety of synthetic transformations. One of the critical steps in photoredox catalysis is the photo-induced electron transfer (PET) from the catalyst in its excited state to the substrate, which usually is the first step of the catalytic cycle. The efficiency of the PET is limited by the diffusional collision of the catalyst and substrate during the lifetime of the excited state of the catalyst.11 This leads to photoredox reactions often suffering from long reaction times, thereby consuming high amounts of light energy, or high catalyst loadings being necessary.1 We herein present a catalyst system, where the PRC is covalently bound to a cavity. This cavity can preassociate the substrate to make the PET more efficient by holding the substrate and catalyst in close proximity (Fig. 1A).12 After completion of the PR catalytic cycle, the catalyst cavity is free to associate with a new substrate. Our catalyst design uses a pillar[5]arene (PA) as the cavity and phenothiazine (PT) as the PRC (Fig. 1B). Pillarenes have become versatile supramolecular hosts due to their facile synthesis, unique pillar shape, the possibility for further functionalization and their host–guest properties.13,14 They have been used in supramolecular catalysis on several occasions15 as well as in photocatalysis.16 While PET to substrates bound by a cage has been reported on a few occasions before,17–20 no application of a pillarene in photoredox catalysis has yet appeared to the best of our knowledge.
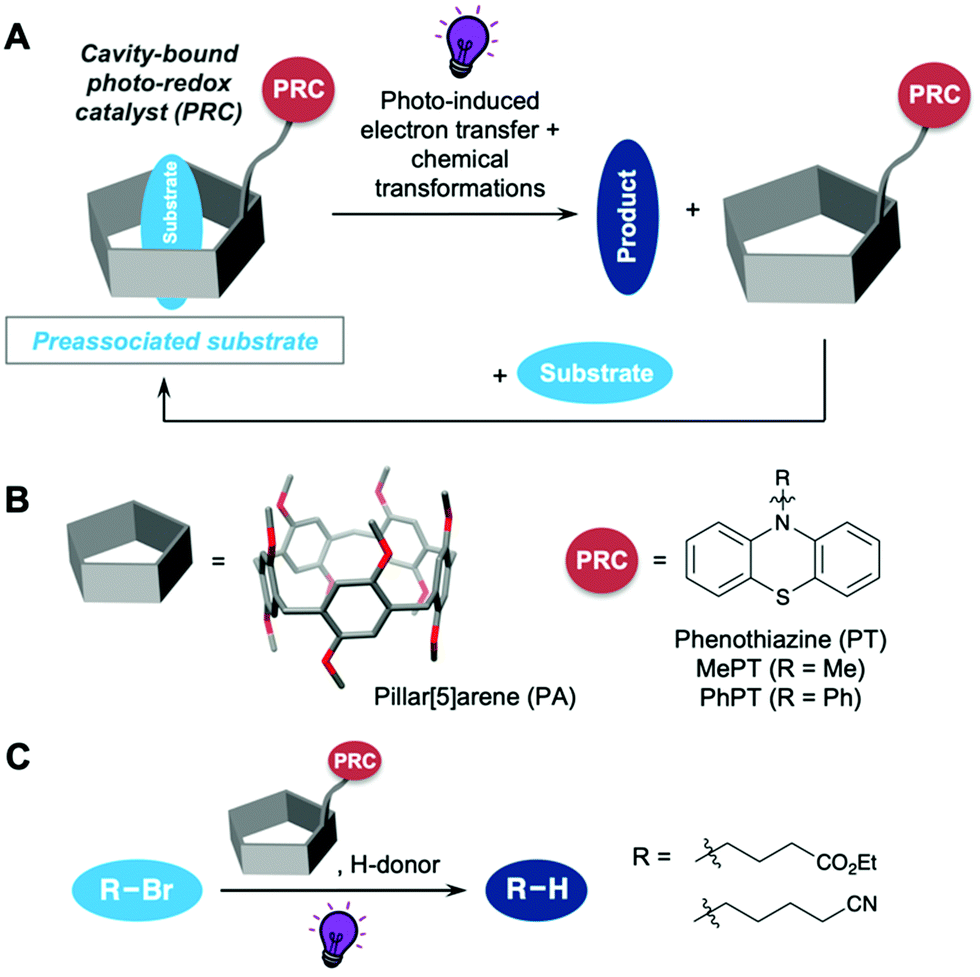 | ||
| Fig. 1 (A) Concept of cavity-promotion in photoredox catalysis, investigated herein; (B) structure of pillar[5]arene (PA), used as a cavity, and phenothiazine (PT) as a photoredox catalyst; (C) photoredox-catalyzed reductive dehalogenation of aliphatic bromides used as a proof-of-concept reaction in this study. | ||
As a proof of concept reaction we chose the reductive dehalogenation of aliphatic bromides (Fig. 1C). This reaction, developed by Hawker and Read de Alaniz in 2015,21 allows for a mild photocatalytic dehalogenation using N-phenyl-PT (PhPT) as the PRC instead of the traditional protocols involving metal–halogen exchange,22,23 tin hydrides24 or other highly toxic reagents.25–27 We used a UV nail dryer lamp as a widely available and cheap light source in this reaction. In spite of its lower light intensity compared to typically used LED setups, we were able to obtain good yields for the reductive dehalogenations, since cavity promotion increased the efficiency of the PET. This is an advantage in comparison to many PRC studies, which use specialized equipment and radiation sources not available to a standard synthetic laboratory. PT derivatives have also been employed in metal-free atom-transfer radical polymerizations28–30 as well as many other reaction types.31,32 Due to their reversible redox chemistry, phenothiazine derivatives have also found use as organic battery materials33–35 and in other supramolecular applications.36
We designed three cavity-linked catalysts PA-PhPT, PA-C4-PhPT and PA-C4-PT (Scheme 1), which differ in the choice of linker between the pillar[5]arene and phenothiazine. The synthesis of all three started from decamethoxy-pillar[5]arene (1),37 which we transformed into mono-hydroxy-PA 2 following literature procedures.38,39 Transformation of the hydroxy to a triflate group followed by Suzuki–Miyaura coupling with PT-based boronic ester 3 led to PA-PhPT in a high yield of 90% over these two steps. For butyl-linked PA-C4-PhPT we reacted 2 with PT-functionalized bromide 4, and nucleophilic substitution furnished the catalyst in excellent yield of 93%. The same procedure using bromide 5 afforded catalyst PA-C4-PT in a high yield of 84%. For the syntheses of PT precursors 3–5, see the ESI.† The PA-linked PRCs were characterized using 1- and 2D 1H- and 13C NMR spectroscopy and HRMS. We determined their excited state reduction potentials using cyclic voltammetry, UV/Vis absorption and emission spectroscopy (for data see the ESI†).8 With E1/2* = −2.84 V (PA-PhPT), −2.85 V (PA-C4-PhPT) and −2.80 V (PA-C4-PT), the cavity-linked phenothiazines have slightly stronger reducing abilities compared to PhPT (E1/2* = −2.56 V) and MePT (E1/2* = −2.69 V, all vs. SCE). Pillarene 1 has no significant absorption at the wavelength used in the reaction (emission maximum of the nail dryer lamp at 365 nm), and hence no excited state of 1 is formed that could interfere with the mechanism.
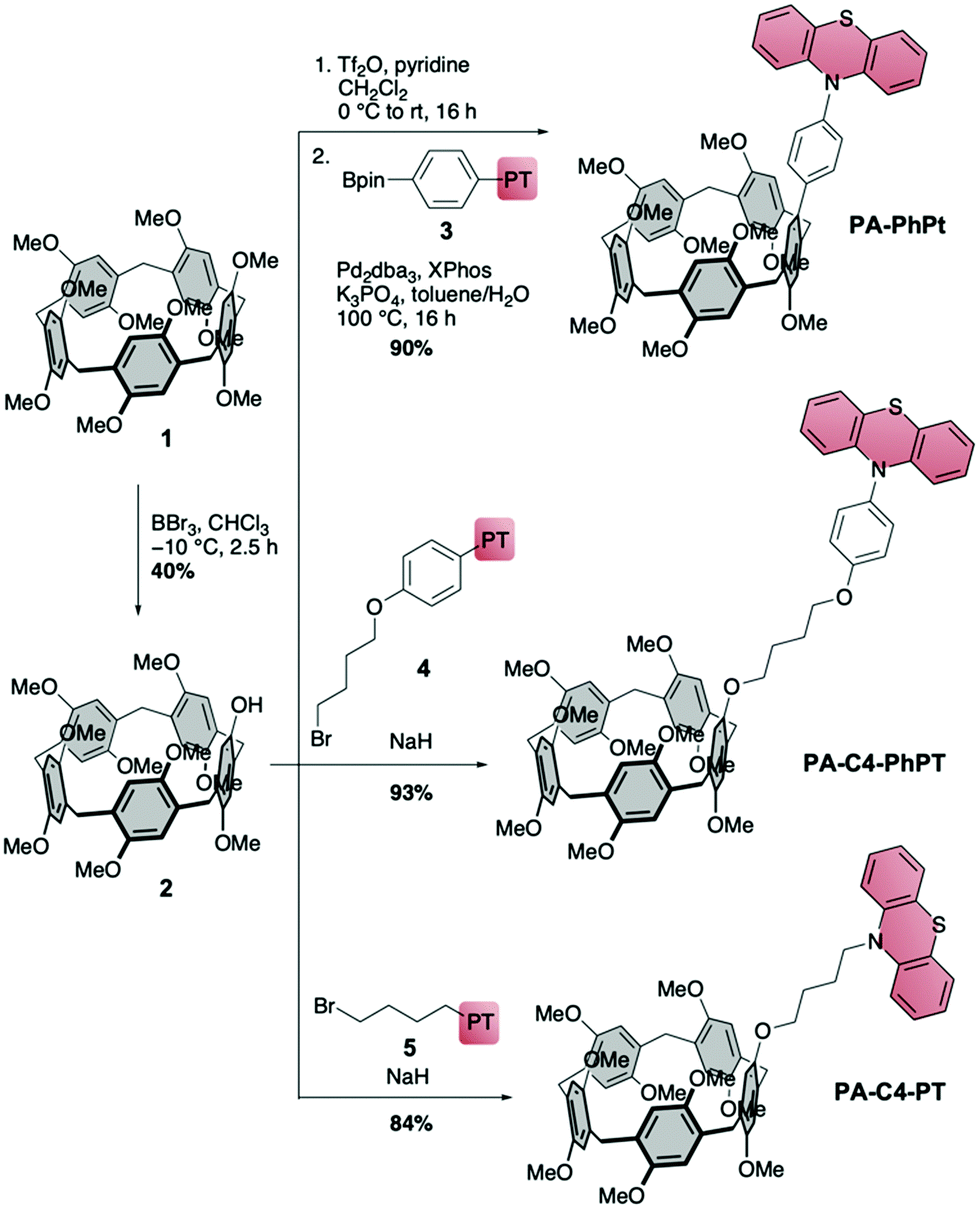 | ||
| Scheme 1 Synthesis of cavity-linked catalysts with pillar[5]arene as the cavity and phenothiazine as a photoredox catalyst (with excited state reduction potentials vs. SCE). | ||
We next employed the cavity-bound PRCs PA-PhPT, PA-C4-PhPT and PA-C4-PT in the reductive dehalogenation of ethyl 4-bromobutanoate (6) and 5-bromopentanenitrile (7) and compared the results to PhPT and MePT (see Fig. 1B) under identical conditions. We used 5 mol% of the PRC and five equivalents each of diisopropylethylamine and formic acid in acetonitrile (Table 1).21 As a light source we used a cheap and widely available nail dryer lamp with an emission maximum of the bulb at 365 nm (for experimental setup see the ESI†). This lamp has a lower photon flux compared to typical LED setups used in the literature, which we determined using ferrioxalate actinometry40 as a simple yet accurate method (see the ESI† for details and comparative values). Hence for the debromination of 6 and 7 using PhPT as PRC, the yield within 48 h amounted to 29% and 28% (entry 1), respectively, while 81% was obtained after 24 h for 6 in the literature using the higher flux LED setup. Using MePT we obtained 17% and 16% yield, respectively, using our setup due to the lower stability of its excited and/or oxidized state compared to PhPT28 (entry 2). Simply adding decamethoxypillar[5]arene (1) to the reaction with MePT did not expedite its course (entry 3), and the presence of sole pillarene 1 did not lead to any transformation (entry 4). Using the cavity-bound PRCs PA-PhPT, PA-C4-PhPT and PA-C4-PT, on the other hand, led to significant increases in yield by factors of 1.6–3.8 compared to PhPT and MePT (entries 5–7). A particularly large improvement is observed for PA-C4-PT with 2.9 and 3.8 times higher yields, respectively, compared to MePT (entry 6); however, this catalyst was slightly less stable towards decomposition compared to the other catalysts due to the alkyl substitution on the phenothiazine nitrogen atom. The best overall performance was observed for PA-C4-PhPT and PA-PhPT with aryl substituents on the phenothiazine nitrogen atoms. Of these two, PA-PhPT is easier to synthesize in a gram scale, since no column chromatography is necessary in the purification of the PT-precursor, and this catalyst could also be recovered quantitatively after each photoredox-catalysed transformation. The increase in reaction rate can also be seen from the kinetic profiles comparing the rates of the dehalogenation of bromopentanenitrile (7) with PA-PhPT and PhPT (see the ESI†).
The results of the photoredox-catalysed experiments demonstrate that the pillarene cavity efficiently expedites the reductive dehalogenation reaction by keeping the substrate in close proximity to the catalyst. This is supported by the known ability of pillar[5]arenes to bind electron-poor alkyl halides.14 The association constant for the complexation of 5-bromopentanenitrile (7) by 1 amounts to 1.7 × 104 M−1.41 This is 30 times higher than that for the debrominated product of the dehalogenation reaction, pentanenitrile, which has an association constant of 5.7 × 102 M−1.41 Hence the reduced product will leave the cavity, thereby releasing the catalyst and making room for a new substrate to enter and undergo the reductive dehalogenation. The calculated structure of the complex between PA-PhPT and 5-bromopentanenitrile (7) in Fig. 2 shows its perfect fit into the cavity and the proximity of the bromide to the PT catalyst. This orientation with the bromide of 7 facing toward the PT catalyst is 5.3 kcal mol−1 more stable than the other orientation where the bromide points downward (see the ESI†).
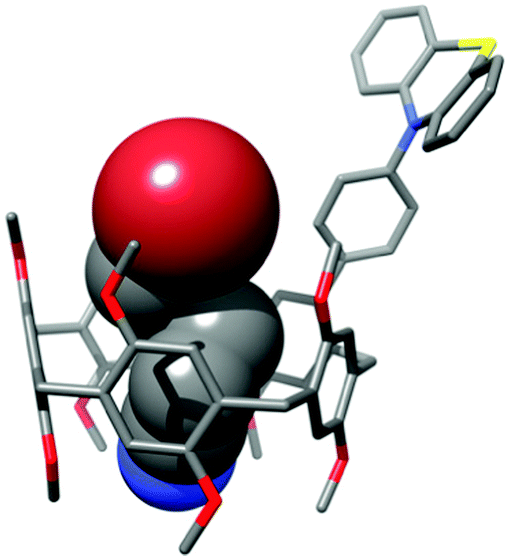 | ||
| Fig. 2 Calculated structure (PBEh-3c) of the complex between PA-PhPT and 5-bromopentanenitrile (7). | ||
Stern–Volmer fluorescence quenching experiments showed that the excited states of the new catalysts PA-PhPT and PA-C4-PhPT are efficiently quenched by the substrates 6 and 7 (see the ESI†). Comparison with non-cavity-bound PhPT revealed a more efficient quenching for PA-PhPT and PA-C4-PhPT than for PhPT due to the substrates binding inside the cavities. Pillarene 1, on the other hand, did not quench the fluorescence of PhPT, demonstrating its innocent nature.
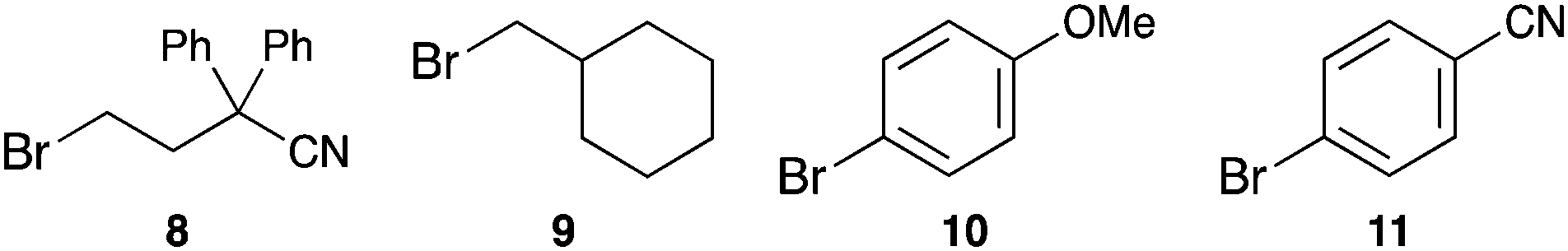
To further prove the postulated concept we performed reactions using four substrates that are sterically hindered and cannot bind inside the pillar[5]arene cavity, namely 4-bromo-2,2-diphenylbutanenitrile (8), (bromomethyl)cyclohexane (9), 4-bromoanisole (10) and 4-bromobenzonitrile (11). We used both the pillarene-bound phenothiazine catalysts PA-PhPT and PA-C4-PhPT as well as PhPT as a cavity-free catalyst (Table 2). In each of these cases we obtained a similar yield for the reductive dehalogenation between the three catalysts (compare entries 1–3 for each substrate in Table 2). The slightly lower yields for PA-PhPT can be rationalized by the lower conformational flexibility of the phenothiazine unit due to the rigid phenylene linker to the pillar[5]arene. These comparative experiments demonstrate that when the substrate is unable to bind inside the cavity, the reaction rate remains similar between cavity- and non-cavity bound photoredox catalysts.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Substrate | |||
| 8 (%) | 9 (%) | 10 (%) | 11 (%) | ||
| a Conversions after 48 h as the mean of three reactions, determined by 1H NMR spectroscopy. | |||||
| 1 | PhPT | 65 | 12 | 51 | 81 |
| 2 | PA-PhPT | 61 | 11 | 50 | 75 |
| 3 | PA-C4-PhPT | 64 | 14 | 50 | 82 |
In conclusion, we introduced a concept in photoredox catalysis where preassociation of the substrate in close proximity to the catalyst makes the reaction significantly more efficient. We synthesized three pillar[5]arene-functionalized phenothiazines PA-PhPT, PA-C4-PhPT and PA-C4-PT as cavity-bound photoredox catalysts and used these in the reductive debromination of ethyl 4-bromobutanoate and 5-bromopentanenitrile. The reactions proceeded with up to 3.8 times higher yield compared with regular phenothiazine catalysts. This concept for photoredox catalysis can be applied to different types and sizes of cavities as well as different catalysts and transformations and will pave the way to making photoredox catalysis more energy- and time-efficient. Furthermore, using cavities that selectively bind one substrate over another would allow realizing substrate selectivity.
Generous support by the German Research foundation and the state of Baden-Württemberg through bwHPC is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- M. H. Shaw, J. Twilton and D. W. C. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- S. G. E. Amos, M. Garreau, L. Buzzetti and J. Waser, Beilstein J. Org. Chem., 2020, 16, 1163–1187 CrossRef CAS PubMed.
- I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- C. Yang, T. Mori, T. Wada and Y. Inoue, New J. Chem., 2007, 31, 697 RSC.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- Y. Wang, G. Ping and C. Li, Chem. Commun., 2016, 52, 9858–9872 RSC.
- K. Wang, J. H. Jordan, K. Velmurugan, X. Tian, M. Zuo, X. Hu and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 9205–9214 CrossRef CAS PubMed.
- H. Qiang, T. Chen, Z. Wang, W. Li, Y. Guo, J. Yang, X. Jia, H. Yang, W. Hu and K. Wen, Chin. Chem. Lett., 2020, 31, 3225–3229 CrossRef CAS.
- R. Gera, A. Das, A. Jha and J. Dasgupta, J. Am. Chem. Soc., 2014, 136, 15909–15912 CrossRef CAS.
- L.-X. Cai, S.-C. Li, D.-N. Yan, L.-P. Zhou, F. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2018, 140, 4869–4876 CrossRef CAS.
- J. R. Piper, L. Cletheroe, C. G. P. Taylor, A. J. Metherell, J. A. Weinstein, I. V. Sazanovich and M. D. Ward, Chem. Commun., 2017, 53, 408–411 RSC.
- J. S. Train, A. B. Wragg, A. J. Auty, A. J. Metherell, D. Chekulaev, C. G. P. Taylor, S. P. Argent, J. A. Weinstein and M. D. Ward, Inorg. Chem., 2019, 58, 2386–2396 CrossRef CAS.
- E. H. Discekici, N. J. Treat, S. O. Poelma, K. M. Mattson, Z. M. Hudson, Y. Luo, C. J. Hawker and J. R. de Alaniz, Chem. Commun., 2015, 51, 11705–11708 RSC.
- P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed.
- W. F. Bailey and J. J. Patricia, J. Organomet. Chem., 1988, 352, 1–46 CrossRef CAS.
- W. P. Neumann, Synthesis, 1987, 665–683 CrossRef CAS.
- A. Krief and A.-M. Laval, Chem. Rev., 1999, 99, 745–778 CrossRef CAS PubMed.
- K. Miura, Y. Ichinose, K. Nozaki, K. Fugami, K. Oshima and K. Utimoto, Bull. Chem. Soc. Jpn., 1989, 62, 143–147 CrossRef CAS.
- M. R. Medeiros, L. N. Schacherer, D. A. Spiegel and J. L. Wood, Org. Lett., 2007, 9, 4427–4429 CrossRef CAS PubMed.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- J. P. Cole, C. R. Federico, C. H. Lim and G. M. Miyake, Aldrichimica Acta, 2019, 52, 7–21 Search PubMed.
- D. A. Corbin, C.-H. Lim and G. M. Miyake, Aldrichimica Acta, 2019, 52, 7–21 Search PubMed.
- F. Speck, D. Rombach and H.-A. Wagenknecht, Beilstein J. Org. Chem., 2019, 15, 52–59 CrossRef CAS PubMed.
- F. Otteny, G. Desmaizieres and B. Esser, in Redox Polymers for Energy and Nanomedicine, ed. N. Casado and D. Mecerreyes, Royal Society of Chemistry, 2020, pp. 166–197 Search PubMed.
- B. Esser, Org. Mater., 2019, 01, 063–070 CrossRef CAS.
- F. Otteny, G. Studer, M. Kolek, P. Bieker, M. Winter and B. Esser, ChemSusChem, 2020, 13, 2232–2238 CrossRef CAS PubMed.
- M. Schmidt, M. Hermann, F. Otteny and B. Esser, Org. Mater., 2020, 02, 235–239 CrossRef CAS.
- T. Boinski and A. Szumna, Tetrahedron, 2012, 68, 9419–9422 CrossRef CAS.
- T. Ogoshi, K. Demachi, K. Kitajima and T. Yamagishi, Chem. Commun., 2011, 47, 7164 RSC.
- Y. Chen, M. He, B. Li, L. Wang, H. Meier and D. Cao, RSC Adv., 2013, 3, 21405 RSC.
- C. G. H. Atchard, Proc. R. Soc. London, Ser. A, 1956, 235, 518–536 Search PubMed.
- X. Shu, W. Chen, D. Hou, Q. Meng, R. Zheng and C. Li, Chem. Commun., 2014, 50, 4820–4823 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthetic manipulations, NMR spectra, cyclic voltammograms, absorption and emission spectra, determination of excited state reduction potentials, determination of photon flux of the nail dryer lamp and details of DFT calculations. See DOI: 10.1039/d1cc03221f |
| This journal is © The Royal Society of Chemistry 2021 |