
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceB–N bond formation through palladium-catalyzed, microwave-assisted cross-coupling of nitrogen compounds with iodo-dodecaborate†
Mahmoud K.
Al-Joumhawy
a,
Tarek
Marei
a,
Akim
Shmalko
 ab,
Paula
Cendoya
a,
Jair
La Borde
a and
Detlef
Gabel
*a
ab,
Paula
Cendoya
a,
Jair
La Borde
a and
Detlef
Gabel
*a
aDepartment of Life Sciences and Chemistry, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany. E-mail: D.Gabel@jacobs-university.de
bA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilov Str., 119991 Moscow, Russia
First published on 13th September 2021
Abstract
Substituted undecahydrido-closo-dodecaborates [B12H11NR2]2− have potential use in materials and drugs, but have presented a synthetic challenge. Microwave-assisted palladium-catalyzed amination of iodo-dodecaborate [B12H11I]2− allows mild and reproducible formation of B–N bonds with aromatic amines, HN-containing heteroaromatics, and amides. The reaction allows general access to amides, reproducible reactions to dodecaborate-substituted anilines, and, for the first time, the substitution of dodecaborate with HN-containing heterocycles.
Boron clusters have gained considerable interest in drug design1,2 and material science.3,4 Icosahedral dicarba-closo-dodecaboranes (carboranes, of which three isomers exist) belong to the most hydrophobic building blocks known. In contrast, dodecaborates (dodecahydrido-closo-dodecaborates) with two negative charges possess good water solubility with alkali counter ions. Nevertheless, they can interact very strongly also with hydrophobic surfaces such as the interior of cyclodextrins5,6 or the exterior of cucurbiturils.7 This unexpected behavior stems in part from the weak coordination of water around the boron clusters.8 This effect has been named superchaotropic.6,9 For making full use of it, dodecaborates should ideally be linked to organic residues exhibiting suitable and complementary characteristics, both in drugs and in materials. While the chemistry for organo-carboranes is well-established, methods for linking the closo-dodecaborates to organic residues are less developed.10 Heteroatoms such as N,11 S,12 and O13 can be introduced and functionalized further by reactions with nucleophiles.14–19
The first B–N derivative of [B12H12]2− was reported by Hertler et al. in 1964.11 This compound, [B12H11NH3]1−, requires purification from di-, and non-substituted clusters. [B12H11NH3]1− offers limited ways for functionalization: it can react with aromatic aldehydes to imines, which can be reduced to benzyl derivatives.20 It can react with carbodiimides to guanidinium derivatives and with aroyl chlorides to amides.21 Alkylation of the N-atom to di- and tri-substituted ammonium salts is possible, but selective monoalkylation could not be achieved.15,16 It can react to a diazonium salt and form azo compounds.22 In nucleophilic aromatic substitution with 1-chloro-2,4-dinitrobenzene, a mixture of two compounds with very poor yield was obtained.18
Direct B–N bond formation of [B12H12]2− with organic nitrogen compounds has been reported, although under harsh conditions and poor selectivity.18,23 For use in the design of drugs and new materials, more general methods for B–N bond formation are desirable. We discovered such a method, using a palladium-catalyzed cross-coupling between iodo-undecahydrido-closo-dodecaborate [B12H11I]2− (1) and a wide range of NH-containing organic compounds, assisted by microwave irradiation. (While previous ways to obtain 1 in pure form required extensive purification, we have recently described a simple and effective way to produce it in pure form.24) Although 1 has been studied in transition-metal-catalyzed reactions,25,26 Pd-catalyzed B–N bond formation in 1 has not been reported. For the formation of a C–N bond, the reaction is known as the Buchwald–Hartwig cross-coupling, and allows a great variety of ligands for successful cross-coupling.27 For the reaction of 1, only one of the tested ligands gives a cross-coupling product, and only with microwave irradiation as heat source.
There is a certain formal analogy between the π aromaticity of benzene and the three-dimensional aromaticity of dodecaborates.28 It has been observed, however, that the chemical reactivity of dodecaborates cannot easily be inferred from that of C-aromatic compounds.29 In the work which we report here, 1 reacts formally similar to an aryl iodide. We found that the reactivity of 1 under Buchwald–Hartwig conditions is, however, very different, and the choice of ligand and heating method are crucial for success.
As model for our initial experiments, we chose the reaction of 1 with aniline (2a). We varied catalytic systems, bases, and reaction conditions. Selected examples are presented in Table 1. We observed dramatic effects of the dialkylphosphine ligand, base, solvent, and heating methods in these initial experiments. To best facilitate the [B12H11I]2− amination, we assessed several Pd/ligand system combinations. Only DavePhos gave the desired aniline derivatives. JohnPhos, XPhos, and t-BuDavePhos gave no reaction of 1, and SPhos and BrettPhos led to consumption of the starting material, but not to a cross-coupling product. JohnPhos and XPhos do not contain other heteroatoms (which potentially might interact with Pd). The two O-containing ligands reacted with 1, but did not lead to any useful yield of the B–N cross-coupling product; rather, [B12H12]2− was obtained. In contrast to DavePhos, t-BuDavePhos did not lead to any consumption of 1, perhaps due to steric reasons.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Ligand | T (°C) | Heating | Time (h) | Consumptiona of 1 (%) | Yielda of 3a (%) |
| Reaction conditions: 1 (1.0 equiv.), aniline (2.0 equiv.), base (2.5 equiv.), Pd2(dba)3 (10 mol%), ligand (20 mol%), solvent (concentration of 1 = 0.1 M). MW: microwave.a Determined from 11B-NMR, see ESI. Ligands: DP = DavePhos, JP = JohnPh, SP = SPhos, tBP = t-BuDavePhos, BP = BrettPhos, XP = XPhos. | ||||||||
| 1 | Cs2CO3 | 1,4-Dioxane | DP | 100 | MW | 0.5 | 20 | <15 |
| 2 | KOt-Bu | 1,4-Dioxane | DP | 100 | MW | 0.5 | 50 | 30 |
| 3 | KOt-Bu | DMF | DP | 150 | MW | 0.5 | 90 | 45 |
| 4 | KOt-Bu | DMF | DP | 150 | MW | 1 | >95 | 45 |
| 5 | KOt-Bu | DMSO | DP | 150 | Oil bath | 48 | 95 | <5 |
| 6 | KOt-Bu | DMSO | DP | 150 | MW | 0.25 | 100 | 65 |
| 7 | KOt-Bu | DMSO | BP | 150 | MW | 0.25 | 90 | <5 |
| 8 | KOt-Bu | DMSO | JP | 150 | MW | 0.25 | 0 | 0 |
| 9 | KOt-Bu | DMSO | tBP | 150 | MW | 0.25 | 0 | 0 |
| 10 | KOt-Bu | DMSO | SP | 150 | MW | 0.25 | 95 | <5 |
| 11 | KOt-Bu | DMSO | XP | 150 | MW | 0.25 | 0 | 0 |
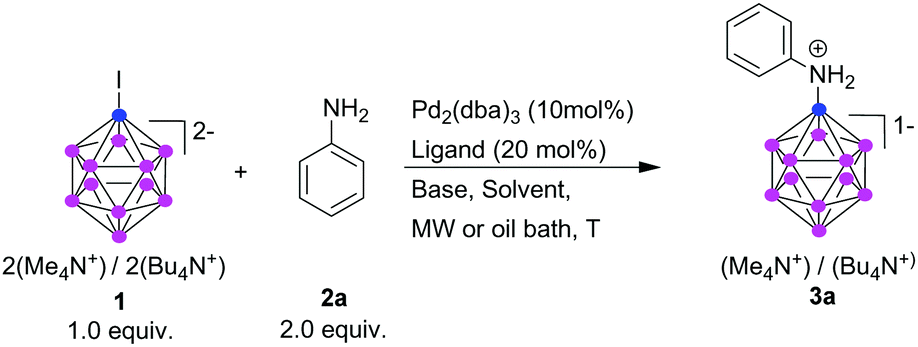
As important as the choice of ligand was the choice of the heating method. Microwave irradiation proved to be far more effective than conventional heating (compare, e.g., entries 5 and 6 of Table 1). Using conventional heating, higher temperatures resulted in higher consumption of 1, but not to 3a; rather [B12H12]2− was formed, with the amination product in less than 10% yield. We therefore replaced conventional heating by microwave irradiation. We had shown before that microwave irradiation greatly increased yield and reduced reaction time in a Sonogashira coupling of 1.30 Also, under microwave irradiation conditions the reaction underwent complete conversion within 15 min with very little side product formation, emphasizing the advantage of using microwave in these types of reactions. Kumada-type cross-coupling with 1 has been reported to proceed by conventional heating, but required longer reaction times.26
Solvents like THF, 1,4-dioxane, toluene, and DME gave low consumption of 1, most likely due to the low solubility of 1, catalyst and/or base, and perhaps due to their lower boiling points or smaller dipole moments (compare entry 2 of Table 1 with entry 6). DMF and DMSO gave the desired aniline derivatives in better yields. Notably, only little of the undesired [B12H12]2− as side product was obtained using DMSO and DMF. DMSO proved to be the most effective solvent, as it could be removed more easily, and was used for further optimization of the B–N coupling reaction conditions.
Also, the choice of base was important. Cs2CO3 (entry 1) was less effective than KOt-Bu (entry 2). KOt-Bu was therefore used for all subsequent reactions. Catalyst loading was important for the yield. Ten mol% of Pd2(dba)3 yielded >65% of the desired product, 5 mol% only between 20–30%. In most cases, we obtained a mixture of the B–N product and [B12H12]2− in a ratio that varied depending on the reaction conditions. In terms of yield, the optimum was obtained with a ratio of 1 to aniline of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.
:2.
Using Pd2(dba)3 (10 mol%), DavePhos as ligand (20 mol%), and KOt-Bu (2.5 equiv.) in DMSO as optimized protocol, we assessed the scope of substituted anilines as coupling partners. For electron-deficient anilines, yields were high (Table 2, 3b, 3c, 3d, 3m, 3n, 3o), except for p-fluoroaniline (3i). p-Phenoxyaniline provided the B–N product in 52% yield (3f), while for the few examples of electron-rich anilines, amination yields were poor (<10%) and hydrogen transfer was dominant, resulting in [B12H12]2− as major product (3g, 3h).
|
|
||||
|---|---|---|---|---|
| Entry | Product 3 | R1, R2, R3 | Consumption of 1a [%] | Yieldb [%] |
| Reaction conditions: 1 (1.0 equiv.), substituted aniline (2.0 equiv.), KO-tBu (2.5 equiv.), Pd2(dba)3 (10 mol%), DavePhos (20 mol%), DMSO (0.1 M).a Consumption of 1 determined by 11B-NMR.b Isolated yield.c Based on 11B-NMR of reaction mixture. | ||||
| 1 | 3a | H, H, H | 85 | 65 |
| 2 | 3b | H, H, NO2 | 100 | 84 |
| 3 | 3c | H, H, CN | 100 | 84 |
| 4 | 3d | H, H, CO2Me | 100 | 81 |
| 5 | 3e | H, H, Me | >95 | 70 |
| 6 | 3f | H, H, OPh | >90 | 52 |
| 7 | 3g | H, H, OEt | >90 | <10 |
| 8 | 3h | H, H, N(Me)2 | >90 | <10 |
| 7 | 3i | H, H, F | >90 | 52 |
| 8 | 3j | H, H, Cl | <5 | Tracec |
| 9 | 3k | H, H, Br | <2 | Tracec |
| 10 | 3l | H, H, I | 0 | 0 |
| 11 | 3m | NO2, H, H | 100 | 83 |
| 12 | 3n | H, NO2, H | 100 | 83 |
| 13 | 3o | NO2, H, NO2 | >95 | 78 |
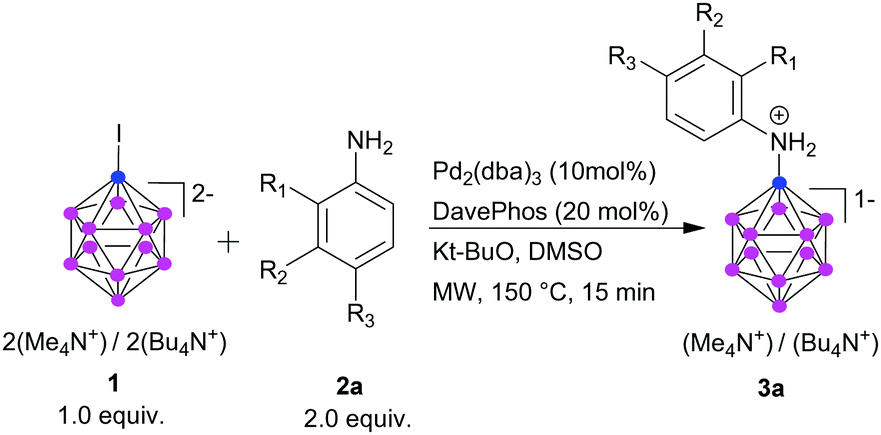
No consumption of 1 was observed with chloro-, bromo- and iodo-aniline under the standard reaction conditions, and the starting material 1 remained untouched. This is possibly due to the faster rate of oxidative addition of palladium with the haloanilines in comparison to that with 1. Collectively, the reaction proceeds well with electron-withdrawing groups in aniline; electron-donating groups react, albeit with lower yield.
Given the promising results achieved, we envisioned that our methodology might also allow us to prepare other types of aromatic primary amines. Coupling of 1-naphthylamine was possible, but only with low yield (Scheme 1). This might be due to steric reasons or to the larger electron density on the N atom.
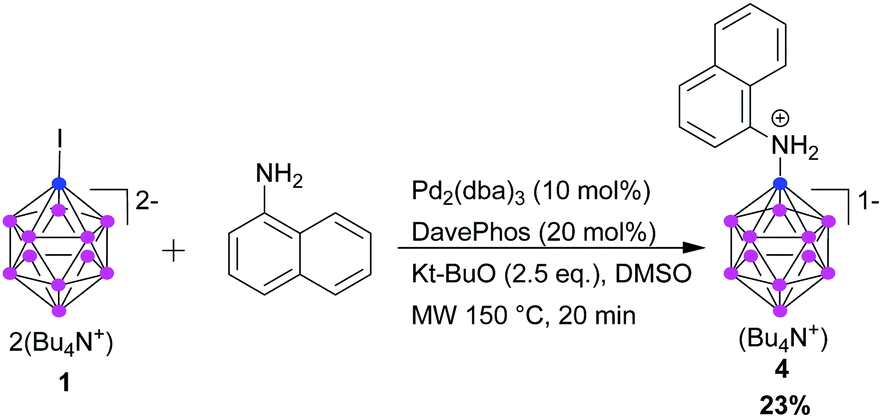 | ||
| Scheme 1 Cross-coupling reaction of 1-naphthylamine with 1. | ||
We examined the coupling of various N-containing heteroaromatics with 1. Such heteroaromatics react in Buchwald–Hartwig reactions.31 The ability to combine aromatic heterocyclic fragments with a boron cluster is an important and challenging task. Heteroaromatic structures are often key fragments of biologically active molecules, and many drugs contain N-heteroaromatics such as pyrrole, imidazole, and indole. A set of aromatic N-heterocycles was tested; the desired B-N products were obtained in excellent yield (Scheme 2, 5a–e). As expected, pyridine did not react.
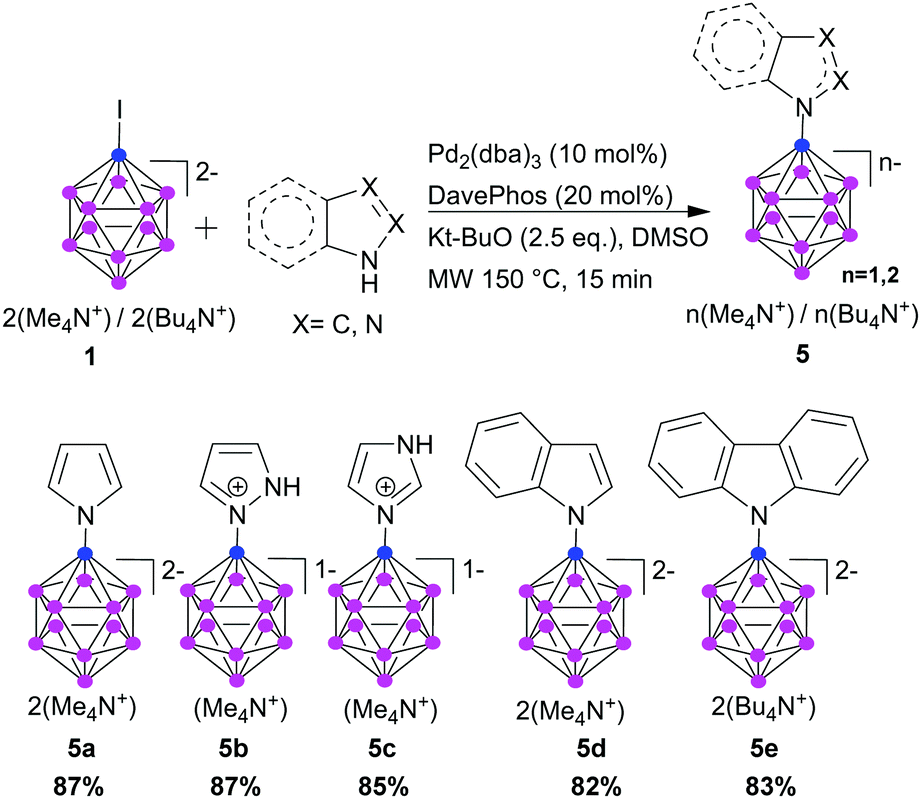 | ||
| Scheme 2 Cross-coupling of 1 with N-containing heteroaromatics. | ||
To expand the applicability of our approach, we investigated the coupling of amides with the dodecaborate cage. We could carry out amidation of 1 under our standard conditions, and the reaction proceeds with aromatic and aliphatic primary and secondary amides, and with sulfonamides (Scheme 3). 2-Nicotinamide and propanamide were successfully coupled in excellent yield. Sulfamide and p-toluene sulfonamide were coupled, however, with moderate isolated yield. The amide coupling products were isolated as protonated species, as described before for the benzoyl derivative.21 We successfully coupled also secondary amides; acetanilide and N-methylacetamide reacted with 1 in excellent yields (6e and 6f, respectively). Also urea could react (6g). 6g can be hydrolyzed to [B12H11NH3]−, offering a new route to this compound without the necessity of purification from non- and di-substituted products found in the present procedure.11
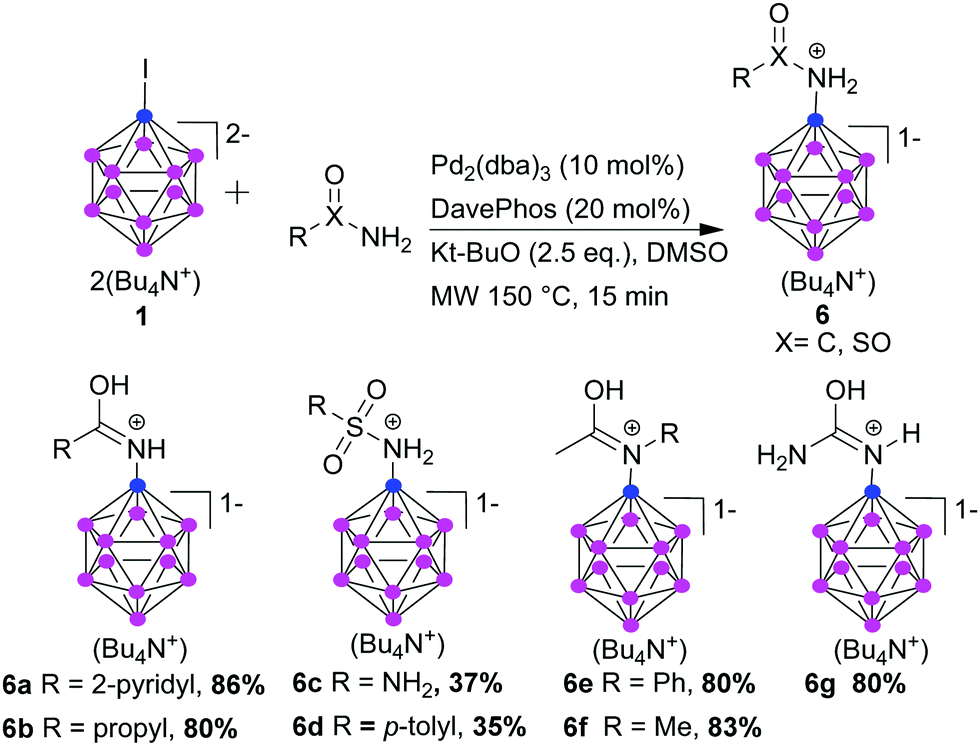 | ||
| Scheme 3 Cross-coupling of 1 with amides. | ||
The reaction that we describe here is possible with aromatic amines, NH-containing heteroaromatics, and amides. For aliphatic amines, our current methodology did not prove effective; rather, we obtained [B12H12]2− as a major product, and the B–N product in single-digit percent yields. We have not yet identified the source of the H atom in any of the reactions, but we suspect that the Pd(II) amine complex undergoes hydride elimination.27
It is also limited to 1. We tried the reaction pf p-nitroaniline with [B12H11Br]2−, the bromine analogue of 1, under conditions that for 1 lead to 3b (see Table 2). No reaction and consequently no formation of 3b was found with[B12H11Br]2−, stressing the connotation that aromatic halides and dodecaborate halides do not react in an identical manner. The limitation to iodine as substituent, and to DavePhos as ligand, is also in contrast to similar reactions on B-halogenated carboranes, where the B–Br bond reacts in cross-coupling reactions, and where other ligands also catalyze the cross-coupling.32,33
In the search for alternative bases, an unexpected B–N bond formation product occurred with DBU. We obtained a product 7 where one of the N atoms of DBU is bonded to a boron atom, a reaction that could be driven to complete conversion and excellent yield by using two equivalents of DBU (Scheme 4).
 | ||
| Scheme 4 Cross-coupling of 1 with DBU. | ||
In summary, we have developed a new Pd-catalyzed cross-coupling of iodo-closo-dodecaborate with aromatic amines, with carboxamides, with NH heteroaromatics, and with sulfonamides. The method provides a new approach to the formation of a B–N bond on dodecaborate. Rapid reaction, high yields, use of readily available catalysts, and reactions with several different nucleophiles that can be coupled to the dodecaborate cage are advantages of this methodology. DavePhos as ligand and microwave irradiation as heating method were essential for successful reaction. Further computational studies on reaction mechanism, and experimental studies to extend the scope of these reactions to include secondary and aliphatic primary amines, are currently underway in our laboratory.
The authors are grateful to the DFG for grant GA 250/55-1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. J. Leśnikowski, J. Med. Chem., 2016, 59, 7738–7758 CrossRef PubMed.
- P. Stockmann, M. Gozzi, R. Kuhnert, M. B. Sárosi and E. Hey-Hawkins, Chem. Soc. Rev., 2019, 48, 3497–3512 RSC.
- B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, New J. Chem., 2011, 35, 1955–1972 RSC.
- R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC.
- K. I. Assaf, D. Gabel, W. Zimmermann and W. M. Nau, Org. Biomol. Chem., 2016, 14, 7702–7706 RSC.
- K. I. Assaf, M. S. Ural, F. Pan, T. Georgiev, S. Simova, K. Rissanen, D. Gabel and W. M. Nau, Angew. Chem., Int. Ed., 2015, 54, 6852–6856 CrossRef CAS PubMed.
- W. Wang, X. Wang, J. Cao, J. Liu, B. Qi, X. Zhou, S. Zhang, D. Gabel, W. M. Nau, K. I. Assaf and H. Zhang, Chem. Commun., 2018, 54, 2098–2101 RSC.
- K. Karki, D. Gabel and D. Roccatano, Inorg. Chem., 2012, 51, 4894–4896 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Angew. Chem., Int. Ed., 2018, 57, 13968–13981 CrossRef CAS PubMed.
- I. Sivaev, V. I. Bregadze and S. Sjöberg, Collect. Czech. Chem. Commun., 2002, 67, 679–727 CrossRef CAS.
- W. R. Hertler and M. S. Raasch, J. Am. Chem. Soc., 1963, 85, 3661–3668 Search PubMed.
- E. I. Tolpin, G. R. Wellum and S. A. Berley, Inorg. Chem., 1978, 17, 2867–2873 CrossRef CAS.
- W. H. Knoth, J. C. Sauer, D. C. England, W. R. Hertler and E. L. Muetterties, J. Am. Chem. Soc., 1964, 86, 3973–3983 CrossRef CAS.
- D. Gabel, D. Moller, S. Harfst, J. Rösler and H. Ketz, Inorg. Chem., 1993, 32, 2276–2278 CrossRef CAS.
- E. Justus, A. Vöge and D. Gabel, Eur. J. Inorg. Chem., 2008, 5245–5250 CrossRef CAS.
- E. Justus, K. Rischka, J. F. Wishart, K. Werner and D. Gabel, Chem. – Eur. J., 2008, 14, 1918–1923 CrossRef CAS PubMed.
- T. Peymann, E. Lork and D. Gabel, Inorg. Chem., 1996, 35, 1355–1360 CrossRef CAS PubMed.
- A. Vöge, E. Lork, B. S. Sesalan and D. Gabel, J. Organomet. Chem., 2009, 694, 1698–1703 CrossRef.
- Y. Zhang, Y. Sun, T. Wang, J. Liu, B. Spingler and S. Duttwyler, Molecules, 2018, 23, 3137 CrossRef PubMed.
- I. B. Sivaev, A. B. Bruskin, V. V. Nesterov, M. Y. Antipin, V. I. Bregadze and S. Sjöberg, Inorg. Chem., 1999, 38, 5887–5893 CrossRef CAS.
- S. Hoffmann, E. Justus, M. Ratajski, E. Lork and D. Gabel, J. Organomet. Chem., 2005, 690, 2757–2760 CrossRef CAS.
- A. R. Genady, Acta Chim. Slov., 2012, 13 Search PubMed.
- T. Koch and W. Preetz, Z. Naturforsch., B: J. Chem. Sci., 1997, 52, 939–942 CAS.
- M. Al-Joumhawy, P. Cendoya, A. Shmalko, T. Marei and D. Gabel, J. Organomet. Chem., 2021, 949, 121967 CrossRef CAS.
- A. A. Kamin and M. A. Juhasz, Inorg. Chem., 2020, 59, 189–192 CrossRef CAS PubMed.
- T. Peymann, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1998, 37, 1544–1548 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed.
- M. Lepšík, M. Srnec, D. Hnyk, B. Grüner, J. Plešek, Z. Havlas and L. Rulíšek, Collect. Czech. Chem. Commun., 2009, 74, 1–27 CrossRef.
- A. Himmelspach, M. Finze, A. Vöge and D. Gabel, Z. Anorg. Allg. Chem., 2012, 638, 512–519 CrossRef CAS.
- G. Mann, J. F. Hartwig, M. S. Driver and C. Fernández-Rivas, J. Am. Chem. Soc., 1998, 120, 827–828 CrossRef CAS.
- R. M. Dziedzic, L. M. A. Saleh, J. C. Axtell, J. L. Martin, S. L. Stevens, A. T. Royappa, A. L. Rheingold and A. M. Spokoyny, J. Am. Chem. Soc., 2016, 138, 9081–9084 CrossRef CAS PubMed.
- R. M. Dziedzic and A. M. Spokoyny, Chem. Commun., 2019, 55, 430–442 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc03215a |
| This journal is © The Royal Society of Chemistry 2021 |