
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast synthesis and redox switching of di- and tetra-substituted bisthioxanthylidene overcrowded alkenes†
Brian P.
Corbet
 ,
Marco B. S.
Wonink
and
Ben L.
Feringa
*
,
Marco B. S.
Wonink
and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, Groningen, 9747 AG, The Netherlands. E-mail: b.l.feringa@rug.nl
First published on 7th July 2021
Abstract
A rapid and efficient method for the synthesis of overcrowded alkenes using (trimethylsilyl)diazomethane provides a range of substituted bisthioxanthylidenes. We show large conformational redox switching from folded to orthogonal states, which tolerates many substitution patterns. The facile access to bisthioxanthylidene switches with the potential for further functionalization, in combination with the reliable redox chemistry, provides major opportunities for the design of electrochemically responsive systems.
Overcrowded alkenes1,2 are an intriguing class of molecules that receive major attention and are particularly attractive in the design of soft actuators3 and responsive materials.4 Our group has a long-vested interest in overcrowded alkenes, for example, towards molecular motors5 and switches, such as bistricyclic aromatic enes (BAEs).6–8 These BAEs are a subset of overcrowded alkenes and include widely studied motifs, such as bifluorenylidenes,9 bis(thio-)xanthylidenes,10 bianthrones11 and their derivatives (Fig. 1a). For various structures in this class of molecules, fascinating photo-, thermo-, mechano-, and electro-chromic behavior as well as dynamic stereochemistry has been demonstrated.2,12 Previously, our group has demonstrated that bisthioxanthylidenes can be used as electrochromic conformational and luminescence switches, both in solution7 and on surfaces.8 Using electrochemical, thermal and photochemical stimuli, multiple states are individually addressable.7 These properties provide attractive possibilities to design functional dynamic systems and responsive materials.
 | ||
| Fig. 1 (a) General structure of bistricyclic aromatic enes (BAEs). (b) Schematic representation of the most common conformers of BAEs. | ||
The characteristic conformational properties found in BAEs, mainly originate from the high steric hindrance around the overcrowded alkene unit.2,12a,b There are two ways these molecules accommodate the overcrowding, which are: twisting around the central 9,9′-axis or folding of the tricyclic units. The twisting and folding can introduce helical chirality to these molecules and is the basis for the four main conformers observed in BAEs (Fig. 1b). The anti- and syn-folded conformers alleviate strain by folding of the tricyclic units, which can lead to pyramidalization of the central olefin carbon atoms. The twisted and orthogonal conformers have two planar tricyclic units and accommodate the overcrowding by rotation around the central double bond axis.
The anti-folded state is the most stable state for neutral bisthioxanthylidenes (Fig. 1, X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) YS). The strain caused by the overcrowding around the central double bond is relieved by a significant folding of the thiopyran motif.2 The syn-folded and the twisted state experience more strain because of a higher overlap between the two halves. Neutral twisted or orthogonal states, with planar halves are possible, however, with significantly higher energies.2 Our group previously demonstrated that, by photo-, electrochemical or thermal switching, higher energy conformers are accessible,4,7 of which the neutral states will rapidly undergo thermal isomerization to the favored anti-folded state. We further showed that a stable, orthogonal state is accessible by oxidation towards a dicationic state with increased single bond character for the central C–C bond and two planar halves.7
YS). The strain caused by the overcrowding around the central double bond is relieved by a significant folding of the thiopyran motif.2 The syn-folded and the twisted state experience more strain because of a higher overlap between the two halves. Neutral twisted or orthogonal states, with planar halves are possible, however, with significantly higher energies.2 Our group previously demonstrated that, by photo-, electrochemical or thermal switching, higher energy conformers are accessible,4,7 of which the neutral states will rapidly undergo thermal isomerization to the favored anti-folded state. We further showed that a stable, orthogonal state is accessible by oxidation towards a dicationic state with increased single bond character for the central C–C bond and two planar halves.7
In the synthesis of overcrowded alkenes (Scheme 1), the most challenging reaction is generally the formation of the overcrowded CC double bond and only a relatively small number of transformations are suitable. Bisthioxanthylidenes are commonly obtained via a Barton–Kellogg synthesis,6a,13 where a diazo compound and a thioketone are used to form olefins via an episulfide intermediate (Scheme 1a). The aforementioned diazo compound is obtained from oxidation of a hydrazone, which in turn is frequently obtained, starting from the corresponding thioketone. Long reaction times of typically 12–16 h are required for each step: the hydrazone formation, coupling and desulfurization. Furthermore, the handling of the often very delicate diazo compound is necessary. Faster and easier synthesis routes, such as the one discussed in this work, can contribute to the study of libraries of compounds and serve as an important addition to the toolbox for incorporation of bisthioxanthylidenes as switches in responsive materials.
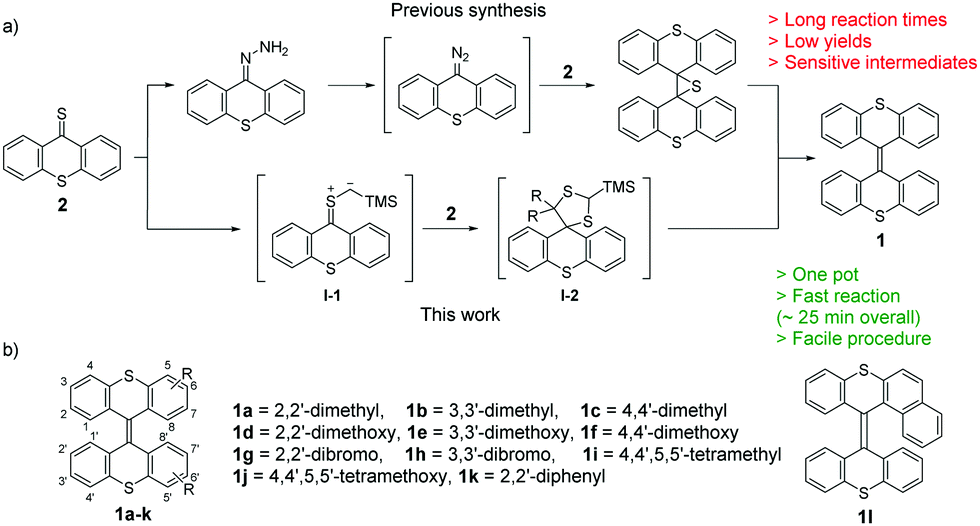 | ||
| Scheme 1 (a) Commonly used previous synthesis route (upper) and novel synthesis route (lower) introduced here, to access bisthioxanthylidenes. (b) Schematic overview of bisthioxanthylidenes (1a–1l) studied. | ||
Here we present an alternative, very facile, synthesis (Scheme 1a) of a variety of substituted bisthioxanthylidenes and their electrochemistry and redox switching (Scheme 1b). By demonstrating a simple, fast synthesis route of substrates with functional handles, in combination with the robust electrochemistry and the intriguing switching modes of bisthioxanthylidenes we expect that these switches will facilitate the development of multi-responsive materials and complex dynamic molecular systems.
Taken inspiration from the recent work of Mlostoń and co-workers,14 we investigated the use of (trimethylsilyl)-diazomethane (TMSCHN2) for the synthesis of bisthioxanthylidenes via dimerization of thioketones. The dimerization reaction cascade likely proceeds via a 1,3-dipolar cycloaddition of TMSCHN2 with thioketone 2, whereupon nitrogen exclusion provides a sulfur ylide (Scheme 1a, I-1). This ylide reacts in another 1,3-dipolar cycloaddition with another equivalent of thioketone 2 leading to a 5-membered dithiane (I-2). The addition of tetrabutyl ammonium fluoride (TBAF) leads to the deprotection of the silyl group, the elimination of methanedithiolate and the formation of the overcrowded alkene 1. By optimization of the reaction conditions for dimerization we developed a fast (∼25 min) and highly efficient method for sterically demanding bisthioxanthylidene formation. One equivalent of the thioketone is treated with TMSCHN2 at −78 °C. No conversion to the sulfonium ylide is observed at this temperature and the reaction mixture is immediately allowed to warm up to room temperature. The conversion of the strongly colored thioketone 2a–k can be easily monitored by TLC analysis or simple observation of the color change. A second equivalent of thioketone was converted within a few minutes after addition. TBAF addition leads to full conversion towards the overcrowded alkene within 5 min. The synthesis of bisthioxanthylidenes from two distinct thioketones is possible. However, this results in a mixture with the homocoupled products (see ESI,† for detailed procedures). The overall time for the complete, one-pot sequence from thioketone to overcrowded olefin is approximately 25 min.
Thioketones 2a–k could be obtained by thionation of corresponding thioxanthones and were submitted to the dimerization conditions immediately after purification. The dimerization reactions show moderate to excellent isolated yields (Scheme 2), while all conversions proceed rapidly and efficiently. It should be noted that the purification of poorly soluble compounds by chromatographic methods is likely the reason for the most significant loss for some compounds showing lower isolated yields. The dimerization reactions were mostly performed on a 0.18–1.26 mmol scale (see ESI†).
 | ||
| Scheme 2 Scope of the dimerization reaction. Compounds 1a and 1d are known in literature,7 compounds 1b, 1c, 1e–1k are novel compounds. For unsymmetrically substituted products only the E-isomer is shown for clarity. | ||
In order to investigate the influence of substitution patterns for bisthioxanthylidenes on electrochemical switching, we synthesized the methyl and methoxy substituted analogues 1a–1f, substituted at the 2,2′-, 3,3′- and 4,4′-positions. The new method was again highly efficient for the synthesis of these compounds and especially methoxy substituted 1d and 1e were obtained in excellent yields. Two bromo substituted bisthioxanthylidenes 1g and 1h were prepared, not only to investigate the substituent effect on switching behavior, but in particular to introduce synthetic handles for the further functionalization of these switches or the incorporation in more complex systems. Starting from unsymmetrically substituted thioketones, the obtained bisthioxanthylidenes products were obtained as E/Z-mixtures with 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 up to 72:28 ratios (see ESI†). The applicability of this method is underlined by the tolerance for various substitution patterns. Further, the methoxy groups also provide a convenient functional handle for further derivatization. The method is also applicable for tetrasubstituted bisthioxanthylidenes, as observed for 1i and 1j, although, for these bisthioxanthylidenes, impurities, which could not be removed by flash column chromatography, were present. In these cases, simple washing steps using Et2O provided the corresponding products with excellent purity.
:50 up to 72:28 ratios (see ESI†). The applicability of this method is underlined by the tolerance for various substitution patterns. Further, the methoxy groups also provide a convenient functional handle for further derivatization. The method is also applicable for tetrasubstituted bisthioxanthylidenes, as observed for 1i and 1j, although, for these bisthioxanthylidenes, impurities, which could not be removed by flash column chromatography, were present. In these cases, simple washing steps using Et2O provided the corresponding products with excellent purity.
The coupling protocol to access sterically demanding bisthioxanthylidenes presented here is a remarkably facile and fast method compared to the previously used Barton-Kellogg reactions. With an efficient method for rapid access to these materials established and a library of compounds (Scheme 1b and Scheme 2, 1a–1l) in hand, we proceeded to investigate their electrochemical properties.
From cyclic voltammetry (Fig. 2b, see ESI,† for cyclic voltammograms of 1a–1l) we observe, in the forward, oxidative direction, a current at Ep,a = 1.15 V vs. saturated calomel electrode (SCE). Initially, 1e is in its anti-folded state and upon oxidation, of overall two electrons, as demonstrated in our previous work,7 a conformational isomerization to the orthogonal dication 1e2+ occurs. The conformational change within the molecule from the anti-folded state with a central double bond proceeds towards an orthogonal state with a central single bond. On the reverse direction, the reduction of 1e2+ is observed at Ep,c = 0.25 V vs. SCE, where the orthogonal 1e2+ accepts two electrons. This initially leads to a twisted state,8 which converts relatively quickly, on the timescale of the electrochemical experiment, towards the thermodynamically favorable anti-folded state. A small oxidation signal, belonging to the twisted state can be observed at 0.34 V vs. SCE. The significant hysteresis between the main oxidation and reduction is caused by the considerable geometrical differences of the anti-folded and orthogonal states. Additional to the different redox response, this geometrical difference aids to explain the differences for the thermal and optical properties.7
 | ||
| Fig. 2 (a) Structure of 1e. (b) Cyclic voltammogram of 1e (1.0 mM), GC, Pt wire, SCE, CH2Cl2, scan speed 10 mV s−1, TBAPF6 (0.1 M). (c) Illustration of the conformational switching, optimized geometry structures obtained from DFT calculations at B3LYP/6-31G** level of theory. | ||
From the observed redox potentials for the synthesized library of compounds (Table 1, see also ESI†), we can see that the electrochemistry of bisthioxanthylidenes is robust and tolerates several substituents and substitution patterns. The presence of electron donating substituents, either in linear or cross conjugated arrangement, with respect to the central olefin, does not lead to a significant difference in redox potentials. The higher electron density of the methoxy substituted bisthioxanthylidenes (1d–1f) has a small effect resulting in a more facile oxidation by stabilization of the dicationic state.
| E p,a (V vs. SCE) | E p,c (V vs. SCE) | Hysteresis (V) | |
|---|---|---|---|
| a 1a–l (1.0 mM), CH2Cl2, TBAPF6 (0.1 M), GC, Pt wire, SCE, 10 mV s−1, room temperature, diffusion limited conditions. b E p,a = anodic peak potential; Ep,c = cathodic peak potential. c For 1h a different electrochemical response is observed (see text). d The first observed wave was selected (see ESI). e Measured as saturated solution because of limited solubility. | |||
| 1a | 1.22 | 0.34 | 0.88 |
| 1b | 1.28 | 0.38 | 0.90 |
| 1c | 1.28 | 0.38 | 0.90 |
| 1d | 1.17 | 0.33 | 0.84 |
| 1e | 1.15 | 0.25 | 0.90 |
| 1f | 1.24 | 0.40 | 0.84 |
| 1g | 1.40 | 0.56 | 0.84 |
| 1h | 1.39 | 0.47d | 0.92d |
| 1i | 1.25 | 0.34 | 0.89 |
| 1j | 1.16 | 0.31 | 0.85 |
| 1k | 1.26 | 0.45 | 0.81 |
| 1l | 1.29 | 0.46 | 0.83 |
As expected, the bromo-substituted bisthioxanthylidenes (1g, 1h) show a significantly higher oxidation potential of 1.40 and 1.39 V vs. SCE, respectively, while still observing the expected geometrical switching. Compound 1g shows the expected redox behavior, only shifted towards more positive potentials, with a hysteresis comparable to the rest of the compounds in the library. However, 3,3′-dibromo-bisthioxanthylidene (1h) shows two closely spaced, significantly smaller reduction signals. This is indicative of a chemical reaction occurring at the dicationic state. We hypothesize that this could be either an oligomerization reaction or nucleophilic attack by water. In 1h, the bromine substituents are conjugated in a para position to the central olefinic bond, which is a plausible explanation for the increased reactivity compared to its regioisomer 1g.
The tetrasubstituted substrates (1i, 1j) show a slight shift towards less positive potentials compared to the disubstituted substrates with a comparable substitution pattern (1c, 1f). Both tetrasubstituted bisthioxanthylidenes show a significantly lower solubility. The number of substituents does not show a significant effect on the electrochemical switching. The presence of aromatic substituents, i.e. diphenyl bisthioxanthylidene (1k), does not interfere with the desired electrochemical switching, with peak potentials at 1.26 and 0.45 V vs. SCE. The hysteresis for 1k (0.81V) is again similar to the hysteresis observed for other substituted bisthioxanthylidenes (Table 1). Finally, the extension of the aromatic core in the non-symmetric 1,2-benzo-compound 1l (Scheme 1b), results in the expected electrochemical switching.
The dicationic and neutral states of bisthioxanthylidenes can be addressed by electrochemical as well as chemical means. Under ambient conditions in the presence of non-dry solvents, compound 1a dissolved in CD3CN (Fig. 3a) was readily oxidized by addition of solid ceric ammonium nitrate (Fig. 3b), leading to a highly selective conversion towards the dicationic 1a2+ with a considerable downfield shift for all signals. Subsequent reduction by addition of solid cobaltocene restores the initial spectrum corresponding to the neutral, folded structure of 1a (Fig. 3c).
 | ||
| Fig. 3 Chemical redox switching. 1H-NMR spectroscopy (500 MHz, CD3CN) of (a) isomerically enriched 1a. (b) 1a2+ obtained by oxidation with ceric ammonium nitrate. (c) 1a, in an equal isomeric ratio, recovered by reduction with cobaltocene. For clarity only the E-isomer of 1a is shown. For further details see ESI.† | ||
We have presented here, a novel, efficient and rapid synthesis method for bisthioxanthylidenes, allowing for various substituent patterns and different regioisomers of these highly overcrowded alkenes. The scope of bisthioxanthylidenes investigated in this study shows that the electrochemical switching is robust and tolerates various substituents. The major geometrically different neutral, anti-folded states and dicationic, orthogonal states are readily addressable both by electrochemical and chemical means. As now easily accessible, reliable and functionalizable switches that can be addressed by multiple stimuli, bisthioxanthylidenes have great potential for their use in molecular switching, for example in electrochromic responsive materials.
This work was supported financially by the European Research Council (ERC; Advanced Grant No. 694345 to B. L. F). We would like to thank Dr R. Costil for fruitful discussions.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- B. L. Feringa and W. R. Browne, Molecular Switches, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed.
- P. U. Biedermann, J. J. Stezowski and I. Agranat, Eur. J. Org. Chem., 2001, 15–34 CrossRef CAS.
- (a) J. Chen, F. K. C. Leung, M. C. A. Stuart, T. Kajitani, T. Fukushima, E. Van Der Giessen and B. L. Feringa, Nat. Chem., 2018, 10, 132–138 CrossRef CAS PubMed; (b) F. K. C. Leung, T. Van Den Enk, T. Kajitani, J. Chen, M. C. A. Stuart, J. Kuipers, T. Fukushima and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 17724–17733 CrossRef CAS PubMed.
- J. Cheng, P. Štacko, P. Rudolf, R. Y. N. Gengler and B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 291–296 CrossRef CAS PubMed.
- (a) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed; (b) S. Kassem, T. Van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC; (c) D. Roke, S. J. Wezenberg and B. L. Feringa, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9423–9431 CrossRef CAS PubMed; (d) J. C. M. Kistemaker, A. S. Lubbe and B. L. Feringa, Mater. Chem. Front., 2021, 5, 2900–2906 RSC; (e) A. S. Lubbe, C. L. G. Stähler and B. L. Feringa, in Out-of-Equilibrium (Supra)molecular Systems and Materials, ed. N. Giuseppone and A. Walther, Wiley-VCH GmbH, 2021, pp. 337–377 Search PubMed.
- (a) W. F. Jager, B. De Lange, A. M. Schoevaars, F. Van Bolhuis and B. L. Feringa, Tetrahedron: Asymmetry, 1993, 4, 1481–1497 CrossRef CAS; (b) B. L. Feringa, W. F. Jager and B. De Lange, Tetrahedron Lett., 1992, 33, 2887–2890 CrossRef CAS; (c) E. M. Geertsema, R. Hoen, A. Meetsma and B. L. Feringa, Eur. J. Org. Chem., 2006, 3596–3605 CrossRef CAS; (d) E. M. Geertsema, A. M. Schoevaars, A. Meetsma and B. L. Feringa, Org. Biomol. Chem., 2006, 4, 4101–4112 RSC; (e) P. M. Erne, P. Štacko, D. J. Van Dijken, J. Chen, M. C. A. Stuart and B. L. Feringa, Chem. Commun., 2016, 52, 11697–11700 RSC; (f) A. C. Coleman, J. M. Beierle, M. C. A. Stuart, B. Maciá, G. Caroli, J. T. Mika, D. J. Van Dijken, J. Chen, W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2011, 6, 547–552 CrossRef CAS PubMed.
- W. R. Browne, M. M. Pollard, B. De Lange, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 12412–12413 CrossRef CAS PubMed.
- O. Ivashenko, H. Logtenberg, J. Areephong, A. C. Coleman, P. V. Wesenhagen, E. M. Geertsema, N. Heureux, B. L. Feringa, P. Rudolf and W. R. Browne, J. Phys. Chem. C, 2011, 115, 22965–22975 CrossRef CAS.
- N. A. Bailey and S. E. Hull, Acta Crystallogr., 1978, 3289–3295 CrossRef CAS.
- (a) J. F. D. Mills and S. C. Nyburg, J. Chem. Soc., 1963, 308–321 RSC; (b) R. Korenstein, K. A. Muszkat and E. Fischer, J. Photochem., 1976, 5, 345–353 CrossRef CAS; (c) G. Sánchez-Sanz, I. Alkorta and J. Elguero, Tetrahedron, 2011, 67, 7316–7320 CrossRef.
- D. H. Evans and R. W. Busch, J. Am. Chem. Soc., 1982, 104, 5057–5062 CrossRef CAS.
- (a) A. Levy, P. U. Biedermann, S. Cohen and I. Agranat, J. Chem. Soc., Perkin Trans. 2, 2001, 2329–2341 RSC; (b) P. U. Biedermann, I. Agranat and J. J. Stezowski, Chem. Commun., 2001, 954–955 RSC; (c) T. Matsue, D. G. Williams and D. H. Evans, J. Electroanal. Chem., 1987, 233, 63–76 CrossRef CAS; (d) X. Yin, J. Z. Low, K. J. Fallon, D. W. Paley and L. M. Campos, Chem. Sci., 2019, 10, 10733–10739 RSC; (e) Y. Ishigaki, T. Hashimoto, K. Sugawara, S. Suzuki and T. Suzuki, Angew. Chem., Int. Ed., 2020, 59, 6581–6584 CrossRef CAS PubMed; (f) Y. Matsuo, Y. Wang, H. Ueno, T. Nakagawa and H. Okada, Angew. Chem., Int. Ed., 2019, 58, 8762–8767 CrossRef CAS PubMed; (g) G. Kortüm, Angew. Chem., 1958, 70, 14–20 CrossRef; (h) E. M. Geertsema, A. Meetsma and B. L. Feringa, Angew. Chem., Int. Ed., 1999, 38, 2738–2741 CrossRef CAS.
- (a) A. Cnossen, J. C. M. Kistemaker, T. Kojima and B. L. Feringa, J. Org. Chem., 2014, 79, 927–935 CrossRef CAS PubMed; (b) D. J. Van Dijken, J. Chen, M. C. A. Stuart, L. Hou and B. L. Feringa, J. Am. Chem. Soc., 2016, 138, 660–669 CrossRef CAS PubMed.
- (a) G. Mlostoń, R. Hamera-Fałdyga and H. Heimgartner, J. Sulfur Chem., 2018, 39, 267–278 CrossRef; (b) G. Mlostoń, P. Pipiak, R. Hamera-Fałdyga and H. Heimgartner, Beilstein J. Org. Chem., 2017, 13, 1900–1906 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc03098a |
| This journal is © The Royal Society of Chemistry 2021 |