
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and enantioselective synthesis of 3-(α-acrylic acid) benzoxaboroles to combat carbapenemase resistance†
You-Cai
Xiao
 a,
Xiao-Pan
Chen
a,
Ji
Deng
a,
Yu-Hang
Yan
a,
Kai-Rong
Zhu
a,
Gen
Li
a,
Jun-Lin
Yu
a,
Jürgen
Brem
b,
Fener
Chen
a,
Christopher J.
Schofield
*b and
Guo-Bo
Li
*a
a,
Xiao-Pan
Chen
a,
Ji
Deng
a,
Yu-Hang
Yan
a,
Kai-Rong
Zhu
a,
Gen
Li
a,
Jun-Lin
Yu
a,
Jürgen
Brem
b,
Fener
Chen
a,
Christopher J.
Schofield
*b and
Guo-Bo
Li
*a
aKey Laboratory of Drug-Targeting and Drug Delivery System of the Ministry of Education and Sichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: liguobo@scu.edu.cn
bDepartment of Chemistry and the Ineos Oxford Institute for Antimicrobial Research, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: christopher.schofield@chem.ox.ac.uk
First published on 14th July 2021
Abstract
Chiral 3-substituted benzoxaboroles were designed as carbapenemase inhibitors and efficiently synthesised via asymmetric Morita–Baylis–Hillman reaction. Some of the benzoxaboroles were potent inhibitors of clinically relevant carbapenemases and restored the activity of meropenem in bacteria harbouring these enzymes. Crystallographic analyses validate the proposed mechanism of binding to carbapenemases, i.e. in a manner relating to their antibiotic substrates. The results illustrate how combining a structure-based design approach with asymmetric catalysis can efficiently lead to potent β-lactamase inhibitors and provide a starting point to develop drugs combatting carbapenemases.
The benzoxaborole ring system has emerged as a useful drug scaffold, due to its intrinsic reactivity, metabolic stability and low toxicity.1 Benzoxaborole derivatives have been developed to treat infectious, inflammatory and neoplastic diseases;2 Tavaborole and Crisaborole are approved for treatment of onychomicosis and mild-to-moderate atopic dermatis, respectively and other benzoxaboroles are in development (Fig. 1a).3 However, most benzoxaborole containing drugs/drug candidates have achiral substituents, hampering the development of selectivity. The development of efficient new methods for the asymmetric construction of chiral benzoxaboroles, including with C3 substituents is a current challenge in the development of boron containing drugs.
 | ||
| Fig. 1 Design of 3-substitued benzoxaboroles as β-lactamase Inhibitors. | ||
Procedures have been recently described for synthesis of C3-substituted benzoxaboroles via reaction of nucleophiles with 2-formyl aryl boronic acid.1a Most, however, suffer from low yields and limited substrate scope.1b Indeed, there has been little progress towards the asymmetric synthesis of 3-substituted benzoxaboroles suitable for biological testing, with only a Wittig/oxa-Michael reaction cascade from 2-formyl aryl boronic acid having been reported.4
A major mechanism of carbapenem resistance involves production of carbapenemases, i.e. evolved β-lactamases that inactivate carbapenem antibiotics, which can be divided into two categories: serine-carbapenemases (e.g. Ambler class A KPC-2 and class D OXA-48 types) and metallo-carbapenemases (e.g. NDM and VIM types).5 With the aim of helping enable agents to overcome carbapenem resistance in Gram-negative pathogens,6 we analysed carbapenem-substrate derived complexes and identified benzoxaboroles with a C3 acrylic acid group as potential inhibitors (Fig. 1b). Here we report use of an asymmetric Morita–Baylis–Hillman (MBH) cascade reaction7 to construct the desired chiral C3-substituted benzoxaboroles. Some of the compounds manifested inhibition of clinically relevant carbapenemases, with the proposed mode of action being validated by crystallography and potentiation of carbapenem activity in cells.
Initially, we utilized 2-formyl-phenylboronic acids as the electrophiles and hexafluoroisopropyl acrylate (HFIPA) as the activated alkene in model reactions. We envisaged challenges relating to the limited substrate scope of the MBH reaction, the desire for asymmetric induction,7c and the presence of a boronic acid group at the benzaldehyde ortho-position. After optimization of the model reaction, we identified reaction conditions using cinchona alkaloid derived chiral tertiary amines C6 and C7 as catalysts (see ESI† for optimization details).
We explored the scope of the asymmetric reaction with various substituted 2-formylphenylboronic acids using C6 (Table 1). The products 3 were hydrolyzed to the corresponding carboxylic acids 4 for biological evaluation. The chiral MBH adducts made using C6 were assigned the R-configuration, based on X-ray analysis of a single crystal of 4a.8
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Timeb (h) | ID, Yieldc (%) | eede (%) |
| a Standard reaction conditions: 1 (0.1 mmol), 2 (0.5 mmol), C6 or C7 (0.01 mmol), Al2O3 (0.4 mmol), 1,4-dioxane (4.0 mL), r.t. b Reaction time for the first step. c Isolated yields over two steps. d ee's were determined by chiral HPLC. e ee after recrystallization from ethyl acetate. f Reaction at 0 °C. | |||||
| 1 | H | H | 29 | (R)-4a, 80 | 90(96) |
| 2 | Me | H | 29 | (R)-4b, 80 | 89 |
| 3 | OMe | H | 43 | (R)-4c, 85 | 90(99) |
| 4 | OEt | H | 10 | (R)-4d, 82 | 86 |
| 5 | OiPr | H | 24 | (R)-4e, 71 | 82 |
| 6 | F | H | 46 | (R)-4f, 86 | 82(90) |
| 7 | Cl | H | 22 | (R)-4g, 70 | 75 |
| 8 | H | Me | 7 | (R)-4h, 84 | 88 |
| 9 | H | OMe | 17 | (R)-4i, 72 | 84 |
| 10 | H | F | 36 | (R)-4j, 91 | 81(96) |
| 11 | H | Cl | 36 | (R)-4k, 81 | 78 |
| 12 | OMe | OMe | 48 | (R)-4l, 84 | 82 |
| 13f | H | H | 96 | (S)-4a, 88 | 60(99.5) |
| 14f | OMe | H | 90 | (S)-4c, 80 | 52(96) |
| 15f | F | H | 76 | (S)-4f, 79 | 48(90) |
| 16f | H | F | 48 | (S)-4j, 65 | 57(95) |

The asymmetric MBH reaction with aromatic aldehydes bearing electron-donating groups is reported to be challenging, owing to their low reactivity.7 However, electron-donating groups, e.g. methyl and alkoxy groups at C4 of the 2-formylphenylboronic ring are well tolerated in our conditions (Table 1, entries 1–5); it is possible that the aldehyde is activated by an intramolecular hydrogen bond between its carbonyl oxygen and the boronic acid, so promoting MBH reaction.9 By contrast, electron-withdrawing substituents at the same position in the phenyl ring delivered relatively low enantioselectivities (entries 6 and 7). Similarly, substituents at C5 of the 2-formylphenylboronic ring were amenable to reaction regardless of the electronic properties of the tested substituents (entries 8–12). However, substituents at either C3- or C6- of the 2-formylphenylboronic ring were not amenable to reaction, with only traces of the desired products being observed, revealing a limitation of the methodology. We applied catalyst C7 for the asymmetric MBH synthesis of (S)-enantiomers; the desired products were observed in good yield, albeit with moderate ee (entries 13–16); pleasingly, excellent ee's were obtained after recrystallization.
We next examined the reaction scope with larger substituents, e.g. aryloxy and benzyloxy groups, at C4 and C5- of 2-formylphenylboronic acids, since structural analyses suggested these derivatives are of interest as β-lactamase inhibitors (representatives are given in Scheme 1).10 Aryloxy substituents, (p-tolyloxy, m-tolyloxy, p-trifluoromethyl-phenoxy) at C4 of 2-formylphenylboronic acids gave products (4m–4o) in high yields and good enantioselectivities. A benzyloxy group at C4 was also amenable to reaction, affording 4p in 81% yield and 87% ee. By contrast, aryloxyl- and benzyloxy substituents at C5 of 2-formylphenylboronic acids gave relatively lower ee‘s compared to the C4 derivatives (4q–4t). Density functional theory (DFT) calculations inform on the mechanism of reaction and rationalise its stereoselectivity (see E. 1 Chemistry in ESI†).
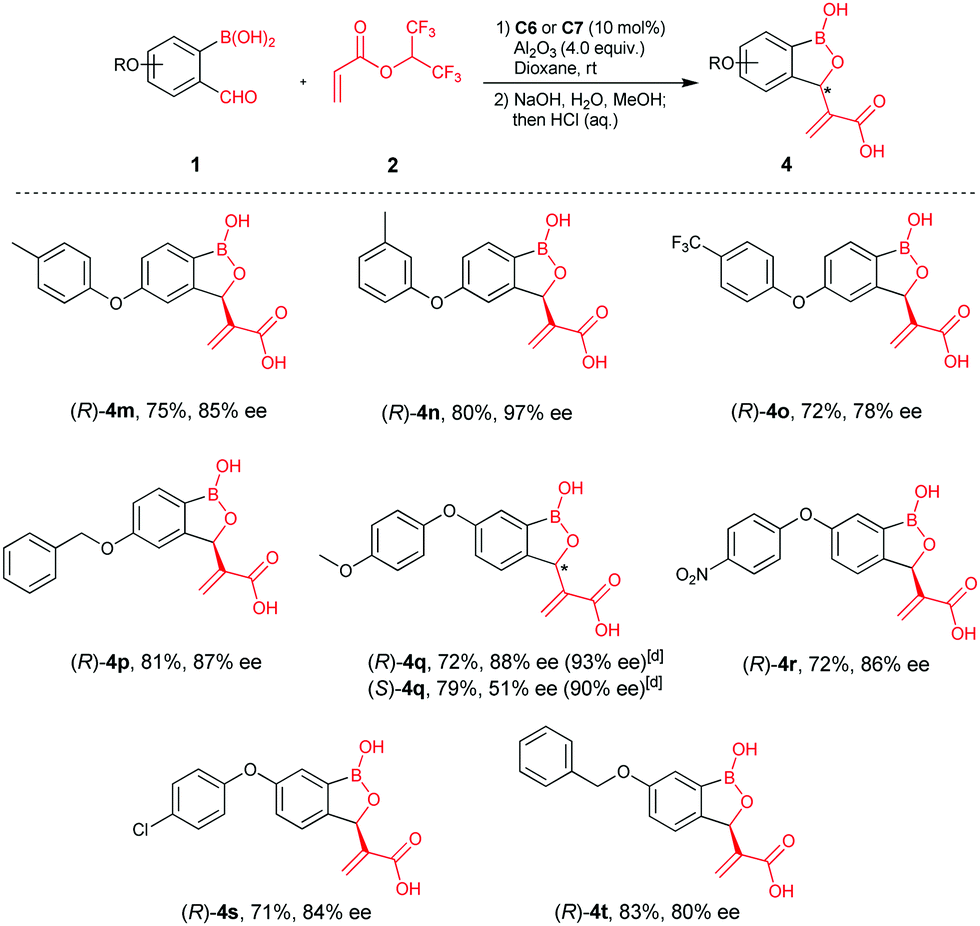 | ||
| Scheme 1 Scope of C3 substituted benzoxaborole synthesis employing aryloxy and benzyloxy substituted 2-formylphenylboronic acids.a,b,c aUnless otherwise stated, reactions were performed with 1 (0.1 mmol), 2 (0.5 mmol), C6 or C7 (0.01 mmol), Al2O3 (0.4 mmol), 1,4-dioxane (4.0 mL), r.t. 12–48 h. bIsolated yields over two steps. cee's determined by chiral HPLC. dee after recrystallization. | ||
We then tested the activity of the benzoxaborole derivatives against purified recombinant forms of 7 types of clinically relevant β-lactamases, i.e. class A SBL KPC-2, class C SBL AmpC, class D SBL OXA-48, class B1 MBLs NDM-1, IMP-1, VIM-1, and VIM-2; the SBL inhibitor avibactam was used as a positive control (Table 2 and Table S1, ESI†).
| Cpd ID | Class A KPC-2 | Class C AmpC | Class D OXA-48 | Class B1 NDM-1 | Class B1 IMP-1 | Class B1 VIM-1 | Class B1 VIM-2 |
|---|---|---|---|---|---|---|---|
| a All tested compounds were recrystallised. IC50 curves are shown in ESI Fig. S1. | |||||||
| (R)-4a | 3.08 | 134.10 | 15.84 | 549.80 | >600 | >600 | 225.40 |
| (S)-4a | 0.48 | 185.7 | 14.06 | >600 | 142.5 | 308.6 | 12.05 |
| (R)-4c | 7.62 | 178.20 | 42.52 | >600 | >600 | >600 | 232.60 |
| (S)-4c | 0.54 | 74.14 | 34.15 | 169.7 | >600 | 466.3 | 22.40 |
| (R)-4f | 3.87 | 140.90 | 10.13 | >600 | 65.65 | 513.8 | 82.80 |
| (S)-4f | 0.34 | 149.8 | 14.68 | 325.3 | 46.5 | >600 | 120.5 |
| (R)-4j | 0.97 | 140.50 | 5.97 | >600 | >600 | >600 | 55.87 |
| (S)-4j | 0.49 | 108.7 | 12.03 | 719.1 | 142.5 | 308.6 | 105.7 |
| (R)-4q | 0.07 | 18.38 | 0.43 | 260.00 | 497.36 | 388.3 | 57.37 |
| (S)-4q | 0.01 | 22.52 | 0.74 | 34.06 | >600 | >600 | 58.49 |
| Avibactam | 0.004 | 0.24 | 0.17 | >600 | 425 | >600 | 19.08 |
The results (Table 2) show both enantiomers of some of the chiral benzoxaborole derivatives manifest moderate, or potent, inhibition of the carbapenemase KPC-2, demonstrating their potential for inhibition of clinically relevant SBLs. In all cases, the (S)-configured compounds (4a, 4c, 4f, 4j, 4q) were more potent than the corresponding (R)-enantiomers, illustrating how chirality can confer selectivity in boron based SBL inhibitors. This selectivity was mostly, but not exclusively observed for the other β-lactamases, substantial inhibition of which was more sporadic than for KPC-2 (Table 2). The KPC-2 stereoselectivity for the benzoxaborole derivatives is interesting since the penicillin substrates of some of the β-lactamases have a (2S) C-3 carbon adjacent to their C-2 carboxylate. The difference in results for the (R)- and (S)-benzoxaboroles may reflect different distances between their carboxylate and electrophilic boron; the analogous carboxylate to β-lactam distance is proposed to be important with respect to antibacterial activity.11
Overall, these results reveal that when combined with an appropriate C-5 or C-6 substituent, the C-3 acrylate substituted benzoxaborole derivatives are potent KPC-2 inhibitors. They were in general much less potent inhibitors for the other tested SBLs/MBLs, but in some cases potent inhibition was observed, suggesting that with optimisation the scaffold could be developed into potent broad spectrum β-lactamase inhibitors.
The abilities of selected Inhibitors to potentiate the antibacterial activity of meropenem (MEM), a clinically important carbapenem, was then investigated for AmpC, TEM-1, or KPC-2 producing clinically derived strains, including Klebsiella Pneumoniae C692, Escherichia coli BAA-2340, and Escherichia coli 11119 (for details, see ESI†). None of these isolates were susceptible to MEM alone (MIC ≥ 16 μg ml−1); for the control strain ATCC25922, the MEM MIC is 0.06 μg ml−1.
Addition of 4a, 4c or 4q at 100 μM substantially reduced the MEM MICs, i.e. by 16-128 fold for the KPC-2 producing E. coli strains; notably, the presence of 10 μM (R)-4a and (S)-4a decreased the MEM MICs to below 2 μg ml−1 for E. coli BAA-2340 and E. coli 11119 (Table 3). Consistent with the biochemical results, reduced (but still clear) activity was observed for AmpC and TEM-1 (both class A SBLs) producing K. pneumoniae C692, i.e. the addition of 100 μM (S)-4a resulted in a 16-fold decrease in the MEM MICs. Overall, consistent with the biochemical results, the (S)-compounds show better cellular activity than the corresponding (R)-enantiomers, illustrating the importance of stereochemistry. Interestingly, although 4q was a better inhibitor than 4aversus against the isolated β-lactmases (Table 2), it was less active in restoring MEM activity in cells (Table 3), possibly reflecting different permeation or efflux properties.
| Compound | Conc. (μM) | MEM (μg ml−1) in presence or absence of inhibitor | ||
|---|---|---|---|---|
| K. pneumoniae C692 (blaAmpC, blaTEM-1) | E. coli BAA-2340 (blaKPC-2) | E. coli 11119 (blaKPC-2) | ||
| a Clinically isolated strains producing KPC-2, AmpC, and/or TEM-1. | ||||
| — | — | >128 | 16 | 64 |
| (R)-4a | 100 | 16 | 0.25 | 0.5 |
| 10 | 128 | 0.5 | 4 | |
| (S)-4a | 100 | 8 | 0.25 | 0.5 |
| 10 | 64 | 0.5 | 2 | |
| (R)-4c | 100 | 128 | 0.5 | 2 |
| 10 | >128 | 2 | 16 | |
| (S)-4c | 100 | 32 | 0.25 | 1 |
| 10 | 128 | 4 | 4 | |
| (R)-4q | 100 | >128 | 1 | 2 |
| 10 | >128 | 2 | 16 | |
| (S)-4q | 100 | 64 | 0.25 | 2 |
| 10 | >128 | 1 | 16 | |
To investigate the structural basis of inhibition by the chiral benzoxaboroles, we obtained KPC-2:(S)-4a and OXA-48:(R)-4a complex structures by co-crystallisation (Table S2, ESI†). The KPC-2: (S)-4a structure reveals a covalent bond between boron and Ser70, with hydrogen bonds between the inhibitor and β3 Thr237, α8 Ser130, β3 Thr235, and β3 Thr237 (Fig. 2a and Fig. S4, ESI†). The OXA-48:(R)-4a structure similarly reveals a tetrahedral boron with hydrogen bonds between the inhibitor α3 Ser70, β5 Tyr211, α5 Ser118, and α10 Arg250; Lys73 of OXA-48 was refined in its catalytically active carbamylated form (Fig. 2b and Fig. S5, ESI†). Superimposition of structures of KPC-2:(S)-4a with KPC-2:reacted imipenem (PDB: 6XJ8),12 and OXA-48:(R)-4a with OXA-48:reacted meropenem (PDB: 6P98)13 reveals that (S)-4a and (R)-4a have binding modes closely related with those of ring-opened products of carbapenems (for more details, see Fig. S7–S9, ESI†). Based on these structures and computational results, it is proposed that the C-6 4-methoxyphenoxyl group of (S)-4q, which occupies a hydrophobic subpocket adjacent to OXA-48 active site, forms an additional hydrogen bond with Asn170 of KPC-2 (Fig. S6, ESI†). This putative interaction may explain why 4q is more potent than 4a and suggests that further optimization of substituents particularly at C-6 should improve the potency of the series for carbapenemase inhibition.
 | ||
| Fig. 2 Crystallographic analyses of the KPC-2:(S)-4a (PDB: 7E9A) and OXA-48:(R)-4a (PDB: 7DML) complexes. | ||
In summary, an enantioselective MBH cascade reaction, employing a bifunctional tertiary amine-carbamate catalyst, was developed for efficient synthesis of 3-substituted benzoxaboroles. The reaction enabled synthesis of C3 acrylate substituted benzoxaboroles in high yield and enantiomeric excess. Screening against clinically relevant SBLs and MBLs, revealed that some of the benzoxaborole derivatives are potent inhibitors of SBLs/MBLs. Crystallographic analyses revealed that active site of (S)-4a to KPC-2 and of (R)-4a to OXA-48 involves reaction with the nucleophilic serine. (R)-4a and (S)-4a potentiate carbapenem activity in cells. The results thus provide an excellent starting point for optimisation to develop selective benzoxaborole derivatives to combat carbapenemase resistance.
The authors thank the staff of BL19U1 beamline of the National Center for Protein Science Shanghai at Shanghai Synchrotron Radiation Facility for assistance during data collection. The authors are grateful to the National Natural Science Foundation (Grant numbers: 81874291, 21907072, and 82073698). CJS thanks the Medical Research Council and the Wellcome Trust for funding. This work was funded in whole, or in part, by the Wellcome Trust (Grant number 106244/Z/14/Z).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews, see: (a) A. Adamczyk-Woźniak, K. M. Borys and A. Sporzyński, Chem. Rev., 2015, 115, 5224 CrossRef PubMed; (b) G. R. Mereddy, A. Chakradhar, R. M. Rutkoski and S. C. Jonnalagadda, J. Organomet. Chem., 2018, 865, 12 CrossRef CAS; (c) Z. J. Leśnikowski, Expert Opin. Drug Discovery, 2016, 11, 569 CrossRef PubMed; (d) C. T. Liu, J. W. Tomsho and S. J. Benkovic, Bioorg. Med. Chem., 2014, 22, 4462 CrossRef CAS PubMed.
- (a) A. Nocentini, C. T. Supuran and J. Y. Winum, Expert Opin. Ther. Pat., 2018, 28, 493 CrossRef CAS; (b) J. Zhang, J. Zhang, G. Hao, W. Xin, F. Yang, M. Zhu and H. Zhou, J. Med. Chem., 2019, 62, 6765 CrossRef CAS PubMed; (c) Y.-H. Yan, Z.-F. Li, X.-L. Ning, J. Deng, J.-L. Yu, Y. Luo, Z. Wang, G. Li, G.-B. Li and Y.-C. Xiao, Bioorg. Med. Chem. Lett., 2021, 41, 127956 CrossRef CAS PubMed.
- J. Plescia and N. Moitessier, Eur. J. Med. Chem., 2020, 195, 112270 CrossRef CAS PubMed.
- G. Hazra, S. Maity, S. Bhowmick and P. Ghorai, Chem. Sci., 2017, 8, 3026 RSC.
- (a) A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503 CrossRef CAS PubMed; (b) A. M. Queenan and K. Bush, Clin. Microbiol. Rev., 2007, 20, 440 CrossRef CAS; (c) N. J. Torelli, A. Akhtar, K. DeFrees, P. Jaishankar, O. A. Pemberton, X. Zhang, C. Johnson, A. R. Renslo and Y. Chen, ACS Infect. Dis., 2019, 5, 1013 CrossRef CAS PubMed; (d) K. Bush and G. A. Jacoby, Antimicrob. Agents Chemother., 2010, 54, 969 CrossRef CAS PubMed.
- (a) K. Bush and P. A. Bradford, Nat. Rev. Microbiol., 2019, 17, 295 CrossRef CAS PubMed; (b) A. Krajnc, P. A. Lang, T. D. Panduwawala, J. Brem and C. J. Schofield, Curr. Opin. Chem. Biol., 2019, 50, 101 CrossRef CAS PubMed; (c) J. Brem, S. S. van Berkel, W. Aik, A. M. Rydzik, M. B. Avison, I. Pettinati, K.-D. Umland, A. Kawamura, J. Spencer, T. D. Claridge, M. A. McDonough and C. J. Schofield, Nat. Chem., 2014, 6, 1084 CrossRef CAS PubMed.
- For selected reviews and examples, see: (a) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed; (b) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS PubMed; (c) D. Basavaiah, K. Venkateswara Rao and R. Jannapu Reddy, Chem. Soc. Rev., 2007, 36, 1581 RSC; (d) H. Pellissier, Tetrahedron, 2017, 73, 2831 CrossRef CAS; (e) J. S. Kumar, C. M. Bashian, M. A. Corsello, S. C. Jonnalagadda and V. R. Mereddy, Tetrahedron Lett., 2010, 51, 4482 CrossRef CAS PubMed.
- Deposition Number 2050141 (for (R)-4a) contains the ESI† crystallographic data for this paper.
- S. Luliński, I. Madura, J. Serwatowski, H. Szatyłowicz and J. Zachara, New J. Chem., 2007, 31, 144 RSC.
- For selected recent reviews, see: (a) Y.-H. Yan, G. Li and G.-B. Li, Med. Res. Rev., 2020, 40, 1558 CrossRef CAS; (b) Y. Xia, K. Cao, Y. Zhou, M. R. K. Alley, F. Rock, M. Mohan, M. Meewan, S. J. Baker, S. Lux, C. Z. Ding, G. Jia, M. Kully and J. J. Plattner, Bioorg. Med. Chem. Lett., 2011, 21, 2533 CrossRef CAS; (c) D. C. McKinney, F. Zhou, C. J. Eyermann, A. D. Ferguson, D. B. Prince, J. Breen, R. A. Giacobbe, S. Lahiri and J. C. Verheijen, ACS Infect. Dis., 2015, 1, 310 CrossRef CAS PubMed.
- N. C. Cohen, J. Med. Chem., 1983, 26, 259 CrossRef CAS PubMed.
- S. C. Mehta, I. M. Furey, O. A. Pemberton, D. M. Boragine, Y. Chen and T. Palzkill, J. Biol. Chem., 2021, 296, 100155 CrossRef CAS PubMed.
- C. A. Smith, N. K. Stewart, M. Toth and S. B. Vakulenko, Antimicrob. Agents Chemother., 2019, 63, e01202 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopy details have been deposited. See DOI: 10.1039/d1cc03026d |
| This journal is © The Royal Society of Chemistry 2021 |