
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible metal-centered reduction empowers a Ni-Corrin to mimic F430†
Christopher
Brenig
a,
Leila
Mosberger
a,
Olivier
Blacque
 a,
Reinhard
Kissner
b and
Felix
Zelder
*a
a,
Reinhard
Kissner
b and
Felix
Zelder
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: felix.zelder@chem.uzh.ch; Web: http://www.felix-zelder.net Fax: +41 44 635 6803
bInstitute of Inorganic Chemistry, ETH Zurich, Vladimir-Prelog-Weg 1-2, 8093, Zurich, Switzerland
First published on 24th June 2021
Abstract
This communication presents a novel truncated NiII-containing metbalamin and describes its reversible one electron reduction to a catalytically active NiI species, that features cofactor F430 model character. Our results strikingly demonstrate that stabilization of NiI is not restricted to the related hydroporhyrinoid ligands and is of relevance to the application of metallocorrins in (biomimetic) catalysis.
Coenzyme F430 is one of the most fascinating transition metal containing cofactors, consisting of a central Ni-ion that is embedded in a macrocyclic corphin ligand (Fig. 1A).1–3 This special porphyrinoid complex is required by the enzyme methyl-coenzyme-M-reductase (MCR) for the last step of bacterial formation of CH4 from CO2.4 In this enzymatic reaction, a thiolate (coenzyme B) and a methyl thioether (methyl-coenzyme M) are coupled to a disulfide (CoM-S-S-Co) under formation of CH4 (Fig. 1B).5 Different mechanisms are controversially discussed for this unusual reaction that has never been observed so far in non-enzymatic chemistry.4,6–9 Important insights regarding the nature, coordination-, and redox chemistry of coenzyme F430 were obtained with synthetic complexes such as a NiII-tetrahydrocorphinate,10 a NiII-tetramethylcyclam11 or a NiII-didehydrocorrin complex embedded in a protein.12 In this context, the structurally closest relationship with the native coenzyme reveals F430M (Fig. 1B), a pentamethylester that is semi-synthetically derived from the natural porphinoid upon dissociation from the protein.13 Electrochemical studies with this hydrophobic model compound strikingly demonstrated one-electron reduction of the inactive NiII form to a catalytically active NiI state.14,15
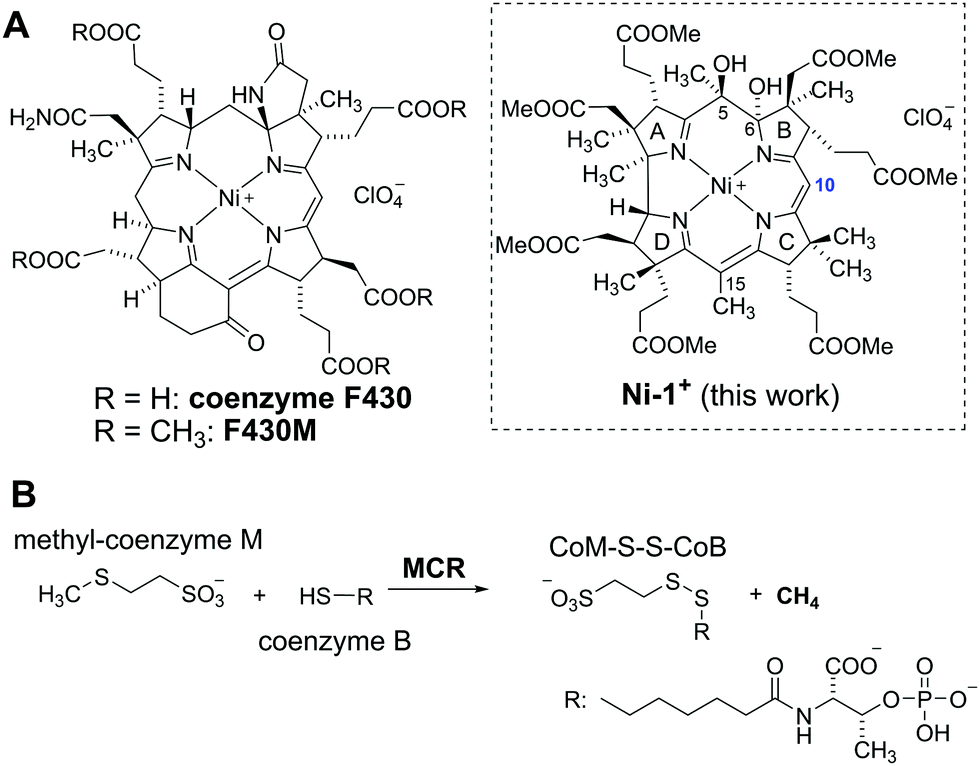 | ||
| Fig. 1 (A) Structures of cofactor F430 and its hydrophobic model F430M in comparison to novel 5,6-dihydroxy-heptamethyl niIIbyrinate (Ni-1+). (B) Last step of methane generation in methanogenic bacteria. | ||
In 2018, we reported an unprecedented chemical pathway for the synthesis of a metbalamin,16i.e. an analogue of vitamin B12 containing other metals than cobalt. In particular, we prepared a 5,6-dihydroxy-niIIbalamin derivative (5,6-DHNibl) in three steps starting from B12, that exhibits an electronic core structure reminiscent of coenzyme F430. Recent DFT studies of Wu and Chen suggest that the NiI-form of a truncated version of our semi-artificial cofactor is enzymatically efficacious and the authors proposed superior MCR activity compared to its natural counterpart.17 In contrast to these encouraging computational results, the redox chemistry and catalytic activity of 5,6-DHNibl and its derivatives, as well of metbalamins in general, has not been studied so far. Inspired by seminal studies with F430M and dicyano-cobester (DCCbs),18–22 the hydrophobic models of F430 and B12, respectively, we report herein on the synthesis of the hydrophobic NiII corrin 5,6-dihydroxy-heptamethyl niIIbyrinate (Ni-1+, Fig. 1) and its reversible one-electron reduction to the catalytically active NiI form (Ni-1), that shares striking key features with reduced forms of F430M (Table 1). Ni-1+ was synthesized as perchlorate salt starting from DCCbs18 in 3 steps (Scheme 1). First, 5,6-dioxo-5,6-seco-Coαβ-dicyano heptamethyl cobyrinate (5,6-DOCbs) was synthesized from DCCbs according to literature.23 Reductive demetallation of 5,6-DOCbs with CoCp2 (2.5 equiv.) and excess KCN in tBuOH led to the formation of metal-free intermediate X that was re-metallated without additional purification with Ni(OAc)2 in MeCN under Ar(g) to the NiII complex 5,6-DONibs in quantitative yields.
| Ni-1+ | F430M | |||
|---|---|---|---|---|
| NiII | NiI | NiII | NiI | |
| a Irreversible. | ||||
| UV/Vis | 277, 447 (MeCN) | 335, 380, 810, 880 (MeCN) | 275, 440 (THF)14 | 278, 382, 754 nm (THF)14 |
| λ max [nm] (solvent) | ||||
| CV | −0.89 (DMF, SHE) | −0.87 (DMF, SHE)14 | ||
| E 1/2 [V] (solvent, ref.) | −0.95 (MeCN, SHE) | −1.15a (MeCN, Fc/Fc+)14 | ||
| EPR (solvent, temp) | Silent | g ⊥ = 1.980, g‖ = 2.194 (MeCN, 110 K) | Silent | g ⊥ = 2.065, g‖ = 2.250 (MeCN, 88 K)14 |
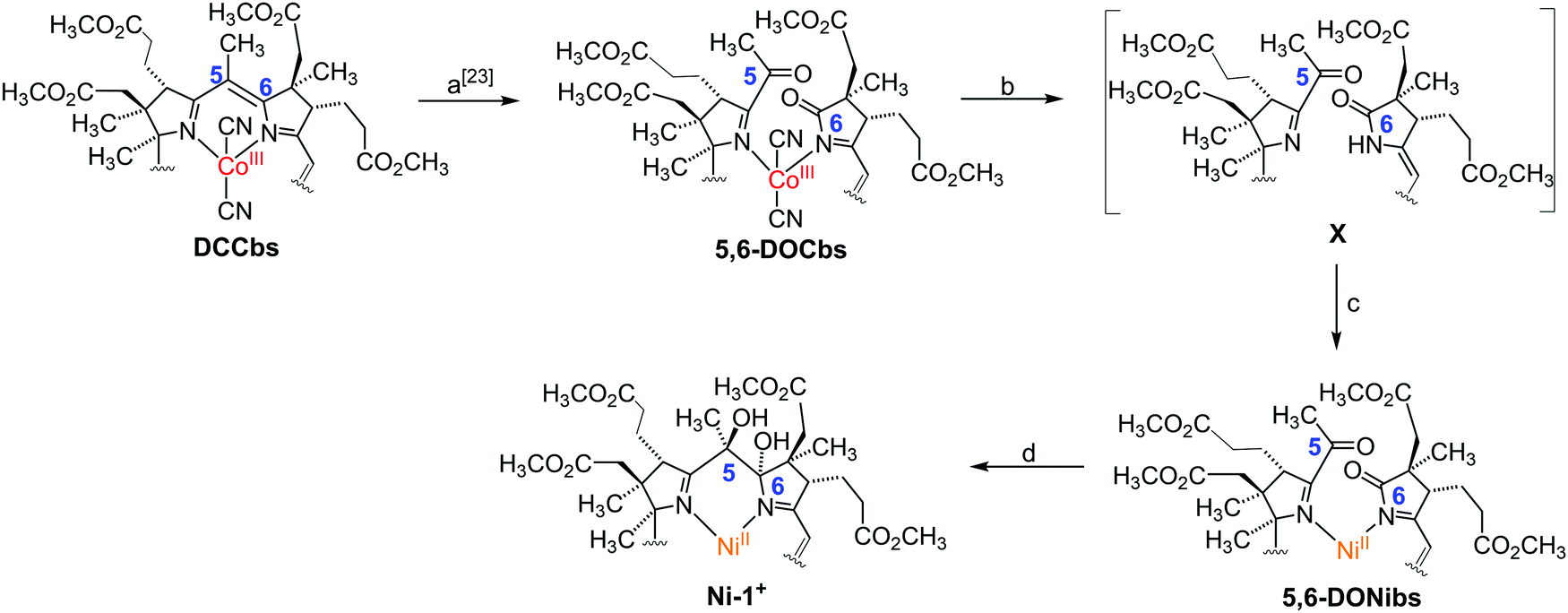 | ||
| Scheme 1 Synthetic route to 5,6-dihydroxy-heptamethyl niIIbyrinate (Ni-1+) starting from DCCbs. Only the ‘northern’ part of the corrin macrocycle is shown, charges and counterions were omitted for clarity. Metal-free intermediate X was not isolated. (a) O2(g), methylene blue, CH3OD, 0 °C, 7.5 h. (b) 1. CoCp2 (2.5 equiv.), 2. KCN (exc.)/H2O, tBuOH, 23 °C, 60 min. (c) Ni(OAc)2·4H2O (10 equiv.), MeCN (anhydr.), 23 °C, 15 min. (d) TiCl4, Zn (s), pyridine, 1,4-dioxane/toluene, 1. 110 °C, 60 min, 2. 23 °C, 90 min. | ||
Notably, isolation of the metal-free intermediate X (Scheme 1) was not successful, due to its apparent high instability. Various protocols were tested to convert 5,6-DONibs to Ni-1+ in a subsequent ring closure reaction through pinacol-type coupling. Unexpectedly, couplings using either CoCp2 or SmI2 failed,16,24 whereas a McMurry type reaction with TiCl4/Zn in 1,4-dioxane finally led to Ni-1+ in satisfactory yield (33%, isolated as Ni-1-ClO4). Integrity of Ni-1+ was verified by thorough spectroscopic investigations using ESI-MS (M+, m/zexp: 1069.61; M+, m/zcalc: 1069.45 for C52H75N4NiO16+), UV/Vis, as well as homo- and heteronuclear one- and two-dimensional NMR spectroscopy. Especially, 1H-NMR studies underscored strikingly that Ni-1+ with a d8-configuration is diamagnetic and hence exhibits a square-planar coordination geometry. The UV/Vis spectrum of Ni-1+ in MeCN displayed maxima at 263, 277, 316 and 447 nm (Fig. S3, ESI†).25 The latter main vis absorption band is shifted towards longer wavelength by 7 nm compared to the vis absorption of F430M, whereas the characteristic double band at 263 and 277 nm in the UV region features higher intensity (εUV/εVIS = 1.33), as it is typical for corrins.26 This is different from the spectrum of F430M that exhibits a less intense single UV band at 275 nm (εUV/εVIS = 0.85).14 Overall the spectrum is similar to that of other NiII-containing corrinoids described earlier by the groups of Eschenmoser26 and Kräutler,27 as well as our group.16 Analysis of the CD spectrum of Ni-1+ indicates that the stereochemistry at C5 and C6 is identical with previously reported 5,6-DHNibl.16 The 1H-NMR spectrum of Ni-1+ in CDCl3 revealed a singlet at 5.57 ppm for the C10 proton (Fig. 1A), thus with a strong highfield shift (Δδ = 0.2 ppm) compared to ring-opened secocorrin 5,6-DONibs. Further details on the NMR structural elucidation of Ni-1+ are reported in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 Cyclic voltammograms of Ni-1+ in DMF showed a distinct cathodic wave with Ep = −(0.93 ± 0.01) V vs. SHE (Fig. 2 and Fig. S15, ESI†) When the scan was reverted at −1.2 V, a distinct anodic wave appeared at Ep = −(0.88 ± 0.01) V with a similar peak current value as the cathodic wave. This observation, complemented by ΔEp ≈ 0.06 V, is consistent with a reversible one-electron transfer redox couple. For the reversible couple, E1/2 = −0.89 V vs. SHE was calculated for Ni-1+ in DMF. A similar single reversible one-electron wave for the Ni-1+/Ni-1 couple was also detected in MeCN at E1/2 = −0.95 V vs. SHE (Fig. S16, ESI†).25 Reduction of the corrin ligand is unlikely to occur above −1.0 V. Free corroles, expected to be reduced more easily, are reported to undergo reduction only at potentials more negative than −1.0 V.28 Therefore, we ascribe the peak pair at −0.89 V to a NiII/NiI redox couple, similar to the one observed for F430M (E1/2 = −0.87 V vs. SHE).14 In a proof of principle study, we investigated the electrocatalytic reduction of alkyl halide 2-chloroacetonitrile (CAN) with electrochemically generated Ni-1. This reaction was extensively studied by Saveant and coworkers with CoIbalamins and CoI-porphyrin complexes.29,30
25 Cyclic voltammograms of Ni-1+ in DMF showed a distinct cathodic wave with Ep = −(0.93 ± 0.01) V vs. SHE (Fig. 2 and Fig. S15, ESI†) When the scan was reverted at −1.2 V, a distinct anodic wave appeared at Ep = −(0.88 ± 0.01) V with a similar peak current value as the cathodic wave. This observation, complemented by ΔEp ≈ 0.06 V, is consistent with a reversible one-electron transfer redox couple. For the reversible couple, E1/2 = −0.89 V vs. SHE was calculated for Ni-1+ in DMF. A similar single reversible one-electron wave for the Ni-1+/Ni-1 couple was also detected in MeCN at E1/2 = −0.95 V vs. SHE (Fig. S16, ESI†).25 Reduction of the corrin ligand is unlikely to occur above −1.0 V. Free corroles, expected to be reduced more easily, are reported to undergo reduction only at potentials more negative than −1.0 V.28 Therefore, we ascribe the peak pair at −0.89 V to a NiII/NiI redox couple, similar to the one observed for F430M (E1/2 = −0.87 V vs. SHE).14 In a proof of principle study, we investigated the electrocatalytic reduction of alkyl halide 2-chloroacetonitrile (CAN) with electrochemically generated Ni-1. This reaction was extensively studied by Saveant and coworkers with CoIbalamins and CoI-porphyrin complexes.29,30
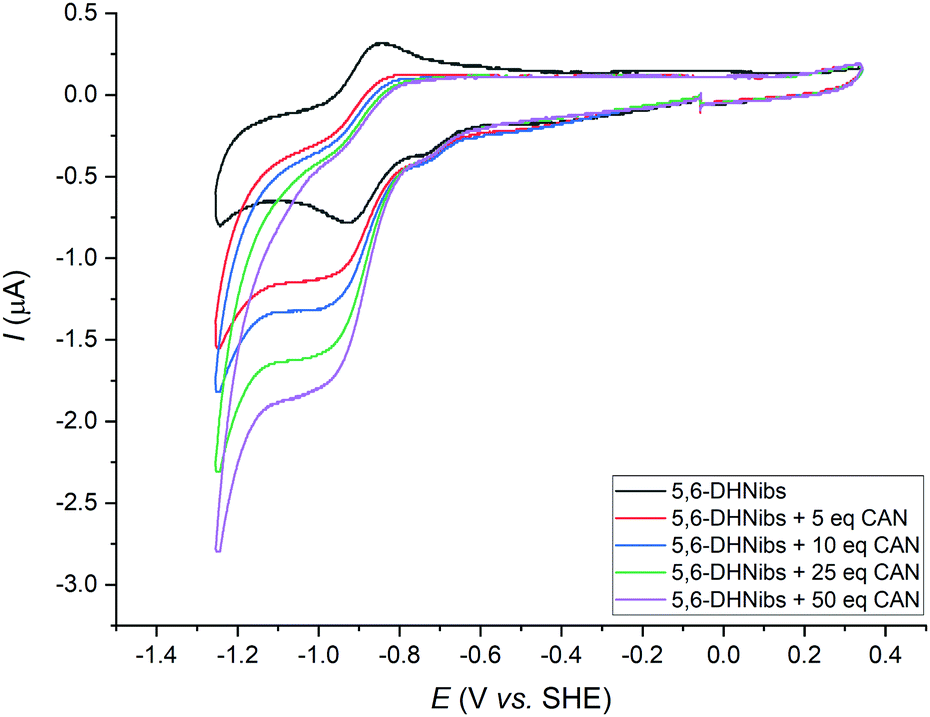 | ||
| Fig. 2 Cyclic voltammograms of Ni-1+ (0.42 mM) in DMF ([TBAPF6] = 0.1 M) in absence (black trace) and presence of chloroacetonitrile (red trace: 5.0 equiv., blue trace: 10 equiv., green trace: 25 equiv. and violet trace: 50 equiv.) Scans from −0.1 V to −1.3 V to 0.3 V and back to −0.1 V at 0.1 V s−1. | ||
Addition of CAN (5.0 equiv.) to a soln. of Ni-1+ (0.42 mM) in DMF and subsequent cyclo-voltammetric scans allowed the detection of a catalytic current at the potential of the first reduction wave (Ep = −0.93 V vs. SHE) as well as a significant decrease of the absolute anodic current. With addition of increasing amounts of CAN (10–50 equiv.), the catalytic current increased notably. Furthermore, the onset potential of the catalytic reaction shifted slightly towards more positive potentials (Fig. 2). Based on these results and earlier studies with F430M31 and NiII-isobacteriochlorins,32,33 we propose reversible one-electron reduction of Ni-1+ to Ni-1 according to eqn (1).
| Ni-1+ + e− → Ni-1 | (1) |
| Ni-1 + NCCH2Cl → [NCCH2–NiIII-1]+ + Cl− | (2) |
| [NCCH2–NiIII-1]+ + e− + H+ → NCCH3 + Ni-1+ | (3) |
It is anticipated that subsequent nucleophilic attack of Ni-1 at the methylene carbon of CAN generates an alkylated NiIII species (eqn (2)),30 that undergoes further reduction and protonation steps to form acetonitrile (NCCH3) and Ni-1+ (eqn (3)). The latter afterwards re-enters the catalytic cycle. While this study demonstrated strikingly that electrocatalysis with Ni-1 is principally possible, detailed electrocatalytic investigations are certainly required to unravel the underlying mechanism and the integrity of the formed product(s), as well as the scope of the biomimetic catalyst, e.g. its potential in the construction of C–C bonds under mild conditions.34 The reduction process was also studied with spectroelectrochemical (SEC) methods. In an SEC cell,25Ni-1+ (2.56 mM in MeCN) showed the expected, reversible reduction wave at −0.9 V (vs. SHE, Fig. S17, ESI†).25 At potentials more negative than −0.6 V, changes in the absorption of Ni-1+ are observed. The main visible absorption band at 447 nm as well absorption at 540, 316, 263 and 240 nm decreases, whereas absorption at 335 and 380 nm increases (Fig. S18, ESI†).25 In addition to these spectroscopic changes, a pronounced strong increase in the NIR region at 810 and 880 nm is detected. These changes are consistent with the spectroscopical behavior of F430M upon electrochemical one-electron reduction to a NiI species for which additional electron density in the dx2−y2 orbital leads to destabilization of corrin π* orbitals due to orbital mixing.14,33 As a direct consequence more energy is required for the π → π* transitions to occur.35 Upon reversal of the applied potential at −1.15 V, changes in absorption between 440 and 880 nm start to reverse, reaching the initial absorbance of Ni-1+ at approx. −0.7 V on the return scan (ΔAbs = 0). The metal-centered nature of the one-electron reduction of Ni-1+ to Ni-1 was further corroborated by EPR spectroscopy.25 Bulk electrolysis in MeCN allowed quantitative conversion of Ni-1+ to Ni-1 at −0.95 V (vs. Ag-wire) within 15 min, as indicated by chronoamperometry. Subsequent EPR measurements in frozen MeCN at 110 K revealed a broad, asymmetric signal with g⊥ = 1.980, g‖ = 2.194 respectively (Fig. S13, ESI†).25 It has a p–p width of 15 mT at 110 K and resembles the spectral feature of singly reduced F430M at 88 K,14 strongly supporting reduction at the metal center to a square-planar NiI species with the additional electron located in the dx2−y2 orbital. Density functional theory (DFT) calculations were conducted to optimize the ground state structures of the Ni-1+ and Ni-1 complexes (for details see ESI†).25 The calculations are consistent with a metal-based reduction of Ni-1+. After reduction, the unpaired electron is mainly located on the metal center as revealed by the Mulliken spin density population at the Ni atom of 0.903 (Fig. 3, right). The population of one virtual frontier orbital by one electron upon reduction gives rise to a more stable structure after relaxation when it occurs on the LUMO+1 orbital (antibonding σ* between the metal dx2−y2 orbital and the nitrogen p orbitals) than on the LUMO (mainly located on the tetradentate ligand with a small participation of the metal, Fig. 3, bottom right). The presence of one electron in the resultant σ orbital induces longer Ni–N bonds in Ni-1 compared to Ni-1+ (+0.068 Å average), but the overall geometry remains very similar with a planar Ni–N4 core.
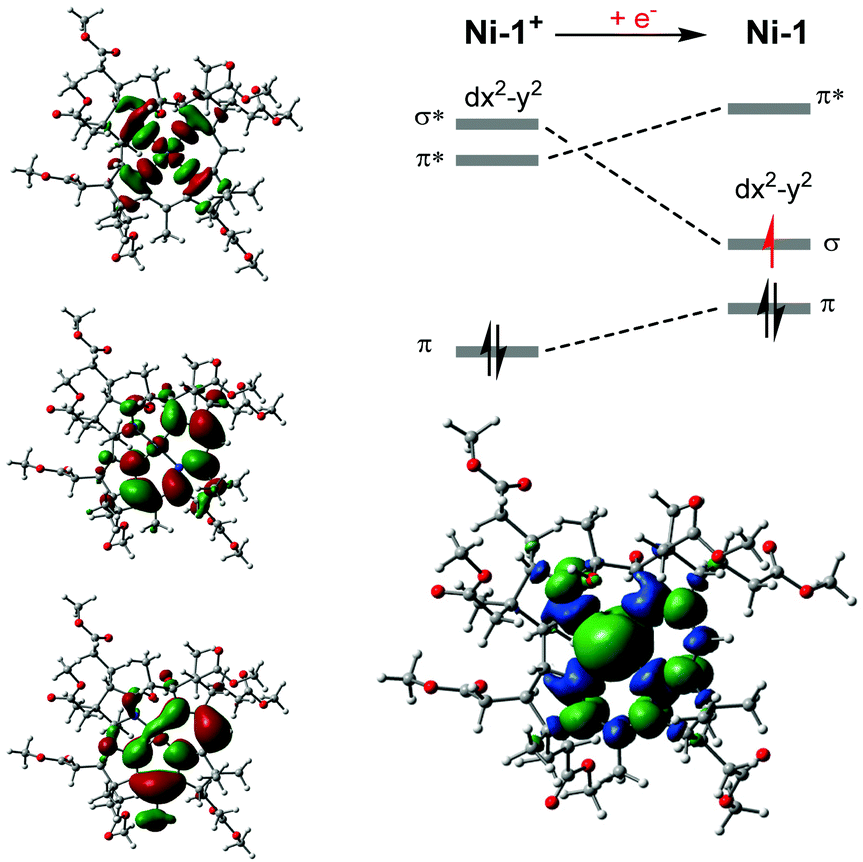 | ||
| Fig. 3 Left: Plots of selected frontier orbitals of Ni-1+; Top, right: Simplified molecular orbital diagram derived from DFT calculations; Bottom, right: Spin density plot of Ni-1. The newly added electron to Ni-1+ to form Ni-1 is highlighted in red. | ||
We have presented a novel truncated NiII-containing metbalamin as a suitable model of cofactor F430. Cyclovoltammetric and spectroelectrochemical studies indicate a reversible one electron reduction to a NiI species, further supported by EPR and computational investigations. A reduction potential of E1/2 = −0.89 V vs. SHE was determined for the NiII/NiI couple of Ni-1+, similar to that of F430M, a superb hydrophobic derivative of the natural Ni-containing coenzyme. In a proof-of-concept study, we demonstrated that electrocatalytic dehalogenation is principally possible with the reduced NiI corrin and intend to extend these studies to biomimetic applications and to the construction of C–C bonds under mild conditions in the near future.
F. Z. and C. B. designed the experiments and wrote the manuscript. C. B., L. M., and R. K. performed the electrochemical measurements and EPR studies. O. B. executed the computational studies.
A generous gift of vitamin B12 from DSM Nutritional Products AG (Basel/Switzerland) is acknowledged. We would like to thank Prof. C. Copéret (ETHZ) for access to his EPR instrument and electrochemical equipment. We are thankful to Dr. Kerstin Oppelt for help with spectroelectrochemical measurements and to Prof. P. Hamm (UZH) for provision of the SEC setup. We acknowledge preparation of synthetic precursors and measurement of IR spectra by Nicolas Yannick Nötel. This work was, in part, financially supported by the UZH ‘Forschungskredit’ (grant-no.: FK-17-088 to C.B.).
Conflicts of interest
There are no conflicts to declare.References
- A. Fässler, A. Kobelt, A. Pfaltz, A. Eschenmoser, C. Bladon, A. R. Battersby and R. K. Thauer, Helv. Chim. Acta, 1985, 68, 2287–2298 CrossRef.
- S. Scheller, M. Goenrich, R. Boecher, R. K. Thauer and B. Jaun, Nature, 2010, 465, 606 CrossRef CAS PubMed.
- S. W. Ragsdale, J. Biol. Chem., 2009, 284, 18571–18575 CrossRef CAS PubMed.
- U. Ermler, W. Grabarse, S. Shima, M. Goubeaud and R. K. Thauer, Science, 1997, 278, 1457–1462 CrossRef CAS PubMed.
- B. Kräutler and B. Jaun, in Concepts and Models in Bioinorganic Chemistry, ed. H.-B. Kraatz and N. Metzler-Nolte, Wiley-VCH, Weinheim, 2006 Search PubMed.
- R. K. Thauer, Biochemistry, 2019, 58, 5198–5220 CrossRef CAS PubMed.
- S. L. Chen, M. R. A. Blomberg and P. E. M. Siegbahn, Chem. – Eur. J., 2012, 18, 6309–6315 CrossRef CAS PubMed.
- T. Wongnate, D. Sliwa, B. Ginovska, D. Smith, M. W. Wolf, N. Lehnert, S. Raugei and S. W. Ragsdale, Science, 2016, 352, 953–958 CrossRef CAS PubMed.
- S. Scheller, M. Goenrich, S. Mayr, R. K. Thauer and B. Jaun, Angew. Chem., Int. Ed., 2010, 49, 8112–8115 CrossRef CAS PubMed.
- C. Kratky, A. Fässler, A. Pfaltz, B. Kräutler, B. Jaun and A. Eschenmoser, J. Chem. Soc., Chem. Commun., 1984, 1368–1371 RSC.
- M. S. Ram, C. G. Riordan, G. P. A. Yap, L. LiableSands, A. L. Rheingold, A. Marchaj and J. R. Norton, J. Am. Chem. Soc., 1997, 119, 1648–1655 CrossRef.
- Y. Miyazaki, K. Oohora and T. Hayashi, J. Organomet. Chem., 2019, 901, 120945 CrossRef CAS.
- A. Pfaltz, B. Jaun, A. Fässler, A. Eschenmoser, R. Jaenchen, H. H. Gilles, G. Diekert and R. K. Thauer, Helv. Chim. Acta, 1982, 65, 828–865 CrossRef CAS.
- B. Jaun and A. Pfaltz, J. Chem. Soc., Chem. Commun., 1986, 1327–1329 RSC.
- B. Jaun, Chimia, 1994, 48, 50–55 CAS.
- C. Brenig, L. Prieto, R. Oetterli and F. Zelder, Angew. Chem., Int. Ed., 2018, 57, 16308–16312 CrossRef CAS PubMed.
- J. Wu and S. L. Chen, Chem. Commun., 2021, 57, 476–479 RSC.
- L. Werthemann, PhD thesis, ETHZ (Switzerland), Zurich, 1968.
- H. Shimakoshi and Y. Hisaeda, Curr. Opin. Electrochem., 2018, 8, 24–30 CrossRef CAS.
- S. M. Chemaly, M. Florczak, H. Dirr and H. M. Marques, Inorg. Chem., 2011, 50, 8719–8727 CrossRef CAS PubMed.
- C. Männel-Croisé and F. Zelder, Anal. Methods, 2012, 4, 2632–2634 RSC.
- C. Männel-Croisé and F. Zelder, Inorg. Chem., 2009, 48, 1272–1274 CrossRef PubMed.
- B. Kräutler, Helv. Chim. Acta, 1982, 65, 1941–1948 CrossRef.
- G. A. Molander and C. Kenny, J. Org. Chem., 1988, 53, 2132–2134 CrossRef CAS.
- ESI†.
- A. Fässler, A. Pfaltz, P. Michael Müller, S. Farooq, C. Kratky, B. Kräutler and A. Eschenmoser, Helv. Chim. Acta, 1982, 65, 812–827 CrossRef.
- C. Kieninger, K. Wurst, M. Podewitz, M. Stanley, E. Deery, A. D. Lawrence, K. R. Liedl, M. J. Warren and B. Kräutler, Angew. Chem., Int. Ed., 2020, 59, 20129–20136 CrossRef CAS PubMed.
- J. Shen, J. Shao, Z. Ou, W. E. B. Koszarna, D. T. Gryko and K. M. Kadish, Inorg. Chem., 2006, 45, 2251–2265 CrossRef CAS PubMed.
- J. E. Argüello, C. Costentin, S. Griveau and J.-M. Savéant, J. Am. Chem. Soc., 2005, 127, 5049–5055 CrossRef PubMed.
- C. Costentin, G. Passard, M. Robert and J.-M. Savéant, Chem. Sci., 2013, 4, 819–823 RSC.
- B. Jaun and A. Pfaltz, J. Chem. Soc., Chem. Commun., 1988, 4, 293–294 RSC.
- M. W. Renner, L. R. Furenlid, K. M. Barkigia, A. Forman, H. K. Shim, D. J. Simpson, K. M. Smith and J. Fajer, J. Am. Chem. Soc., 1991, 113, 6891–6898 CrossRef CAS.
- A. M. Stolzenberg and M. T. Stershic, J. Am. Chem. Soc., 1988, 110, 6391–6402 CrossRef CAS.
- A. Wuttig, J. S. Derrick, M. Loipersberger, A. Snider, M. Head-Gordon, C. J. Chang and F. D. Toste, J. Am. Chem. Soc., 2021, 143, 6990–7001 CrossRef CAS PubMed.
- M. W. Renner, L. R. Furenlid and A. M. Stolzenberg, J. Am. Chem. Soc., 1995, 117, 293–300 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc02945b |
| This journal is © The Royal Society of Chemistry 2021 |