
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cluster expansion and vertex substitution pathways in nickel germanide Zintl clusters†
Oliver P. E.
Townrow
 a,
Andrew S.
Weller
*b and
Jose M.
Goicoechea
*a
a,
Andrew S.
Weller
*b and
Jose M.
Goicoechea
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: jose.goicoechea@chem.ox.ac.uk
bDepartment of Chemistry, University of York, YO10 5DD, UK. E-mail: andrew.weller@york.ac.uk
First published on 22nd June 2021
Abstract
We describe the reactivity of the hypersilyl-functionalized Zintl cluster salt K[Ge9(Hyp)3] towards the nickel reagents Ni(COD)2 and Ni(Cp)2, which gives rise to markedly different complexes. In the case of Ni(COD)2 (COD = 1,5-cyclooctadiene), a dianionic sandwich-like cluster [Ni{Ge9(Hyp)3}2]2− (1) was obtained, in line with a simple ligand substitution reaction of COD by [Ge9(Hyp)3]−. By contrast, when an analogous reaction with Ni(Cp)2 (Cp = cyclopentadienyl) was performed, vertex substitution of the [Ge9(Hyp)3]− precursor was observed, giving rise to the nine-vertex nido-cluster (Cp)Ni[Ge8(Hyp)3] (2). This is the first instance of vertex substitution at a hypersilyl-functionalized Zintl cluster cage. The electrochemical behavior of these compounds was explored and showed reversible redox behaviour for both clusters.
Intermetallic materials consisting of nickel and germanium (nickel germanides) have been recently explored as catalysts,1 components in microelectronics,2 and thermoelectric materials.3 Methods for the preparation of such compounds include atomic layer deposition and direct current (DC) or radio frequency (RF) sputtering, which allow for the preparation of high purity samples. Given the important technological applications of such materials, the development of alternative synthetic techniques based on well-defined molecular precursors is an attractive prospect. In this context, one such family of compounds that are of interest are heteroatomic Zintl clusters composed of main group and transition metal elements.4 These soluble molecular species can be viewed as mimics of binary intermetallic compounds.5 As far as nickel germanide clusters are concerned, an array of species with varying compositions have been isolated to date such as [Ni@Ge9]3−,6 [Ni@Ge9Ni(CO)]3−,6 [Ni3@(Ge9)2]4−,7 and [Ge5Ni2(CO)3]2−.8 The formation of many of such clusters, [Ni2@Ge14Ni4(CO)5]4− for example,9 involves cluster-fragmentation pathways that are poorly understood owing to the lack of suitable spectroscopic handles to monitor such reactions. This aspect of Zintl cluster chemistry – the mechanisms by which an otherwise robust molecular precursor, such as the nonagermanide tetra-anion, [Ge9]4−, redistributes in solution to afford higher nuclearity clusters – remains for the most part a mystery, although recently some studies have aimed to elucidate viable pathways.10
In an effort to probe such reactivity, we have recently become interested in studying the tris-functionalised nonagermanide cluster [Ge9(Hyp)3]− (Hyp = Si(SiMe3)3).11 Over the last ten years this species has received significant attention as a supporting ligand for transition metals.12–15 For example, we recently reported its use in the synthesis of a novel Rh-based homogeneous catalyst for the hydrogenation of cyclic alkenes.15 The principal advantages of [Ge9(Hyp)3]− as a ligand are that it is (highly) soluble in non-polar aprotic solvents, and that reactions can be studied in greater detail using solution NMR spectroscopic techniques given the presence of 1H and 29Si nuclei in the hypersilyl substituents. A number of metal-containing clusters have been isolated containing this cluster as a support, that exhibit a variety of coordination modes (η1, η3, η4, η5), which depend on the electronic requirement of the metal centres in question.12–15 Empirical observations also suggest that the [Ge9(Hyp)3]− precursor is less prone to fragmentation than that of its unsubstituted counterpart [Ge9]4−.4,16
In addition to cluster expansion reactions, whereby higher nuclearity clusters form on reaction of Zintl clusters with transition metal reagents, another interesting class of reactions are those in which a transition metal fragment replaces one of the cluster vertices, so-called vertex substitution reactions.17 It has been postulated that vertex substitution of [E9]4− clusters (E = Ge, Sn, Pb) may be an early step towards cluster expansion, whereby the fragmented cluster combines with another to form higher nuclearity cages.10,18 Some examples of clusters which have undergone vertex substitution reactions include [(Cp)TiSn8]3− and [(CO)3FeGe8]3−,18,19 both of which exhibit cluster topologies encountered in higher nuclearity clusters such as [Ni2@Sn17]4−.20 The mechanisms involved in these processes remain poorly understood, largely because of the lack of methods to study the solution behaviour. Establishing complementary reactivity in their functionalized, more soluble counterparts may allow us to monitor these processes.
Herein, we report the reactions of homoleptic nickel organometallics Ni(COD)2 (COD = 1,5-cyclooctadiene) and Ni(Cp)2 (Cp = cyclopentadienyl), both well studied precursors and dopants for Ni containing materials,21,22 with K[Ge9(Hyp)3]. These studies show that the nature of the nickel precursor has a significant effect on the clusters formed in solution. Using Ni(COD)2 results in cluster expansion, while in contrast Ni(Cp)2 replaces one of the germanium vertices of the [Ge9(Hyp)3]− cluster, in a vertex substitution reaction.
The reaction of Ni(COD)2 and two equivalents of K[Ge9(Hyp)3] in benzene or toluene results in the formation of a dark green solution, presumably of the complex K2[Ni{Ge9(Hyp)3}2] (K2[1]).‡ This product is unstable in solution for more than 2 days and also if placed under vacuum, forming a brown suspension from which the previously reported [K(tol)3][Ge9(Hyp)3]·tol was recovered from toluene.23 However, addition of 2,2,2-crypt (4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]-hexacosane) to the reaction mixture allows for the isolation of the dianionic formally Ni(0) sandwich compound [K(2,2,2-crypt)]2[Ni{Ge9(Hyp)3}2] ([K(2,2,2-crypt)]2[1]), which is sparingly soluble in THF (Scheme 1).
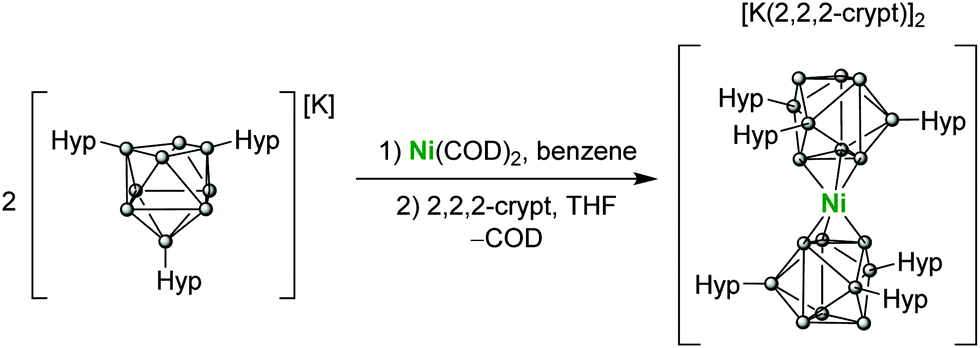 | ||
| Scheme 1 Preparation of [K(2,2,2-crypt)]2[Ni{Ge9(Hyp)3}2] ([K(2,2,2-crypt)]2[1]). | ||
Dark green-blue crystals suitable for single crystal X-ray crystallography were grown from a concentrated THF solution at −40 °C. The cluster adopts a D3d symmetric sandwich-like geometry in which the central Ni atom is coordinated by a triangular face of each of the two flanking clusters (Fig. 1). The coordinated Ge–Ge distances are elongated by ∼0.2 Å compared with the non-coordinated face of the cluster. The dianionic cluster 1 is valence isoelectronic with related clusters such [M[Ge9(Hyp)3]2] (M = Zn–Hg) and [Au[Ge9(Hyp)3]2]−.13i,j There is also a close structural relationship to the substituent-free species [Ni3@(Ge9)2]4−,7 although it is notable that the Ni–Ge bond lengths in 1 are ca. 0.1 Å shorter, 2.436(1)–2.443(1) Å, (cf. 2.505(1)–2.540(1) Å in [Ni3@(Ge9)2]4−), which is presumably due to the absence of interstitial nickel atoms in the former, and its reduced overall charge.
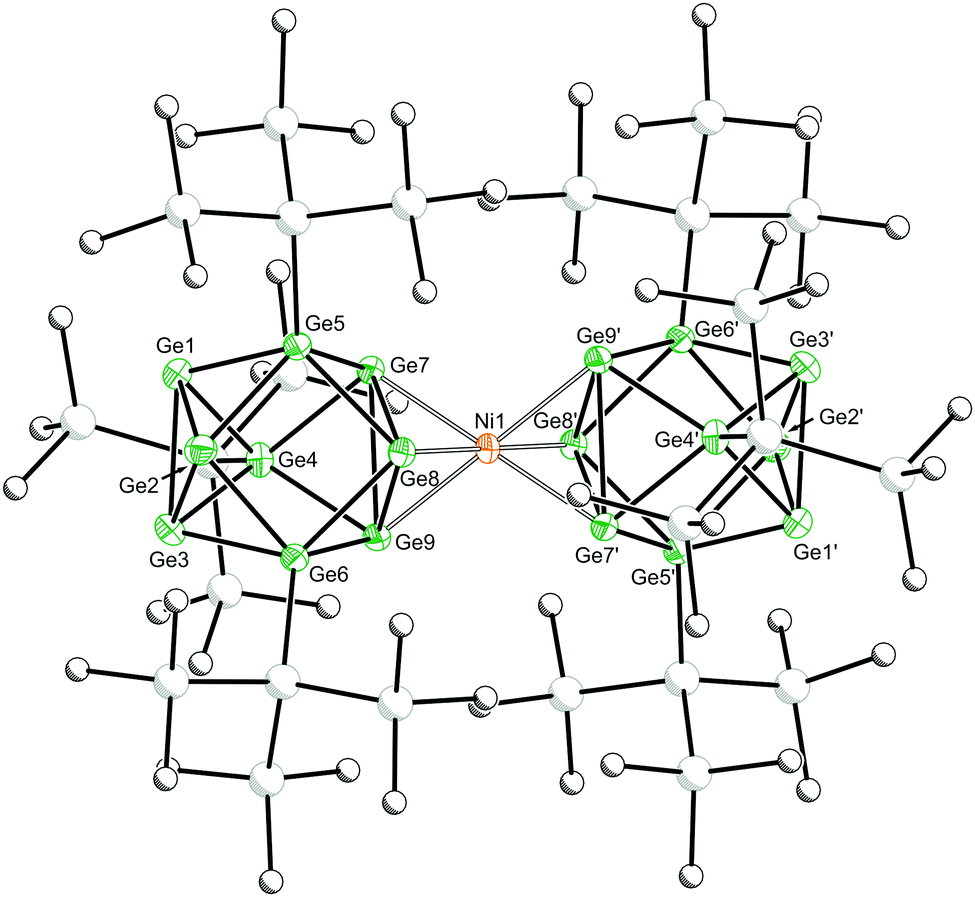 | ||
| Fig. 1 Molecular structure of [K(2,2,2-crypt)]2[1]·2THF. Anisotropic displacement ellipsoids are set at 50% probability. Hydrogen atoms, [K(2,2,2-crypt)]+, and solvent of crystallisation have been omitted for clarity. Carbon and silicon atoms are pictured as spheres of arbitrary radii. Selected bond distances [Å]: Ni1–Ge7:2.442(1), Ni1–Ge8:2.436(1), Ni1–Ge9:2.443(1), Ge1–Ge2:2.679(1), Ge1–Ge3:2.643(1), Ge2–Ge3:2.631(1), Ge7–Ge8:2.900(1), Ge7–Ge9:2.830(1), Ge8–Ge9:2.826(1). Symmetry operation ’: 1 − x, 1 − y, −z. | ||
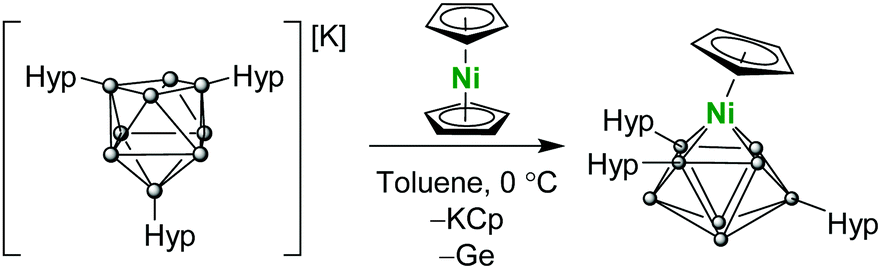 | ||
| Scheme 2 Preparation of (Cp)Ni[Ge8(Hyp)3] (2). | ||
NMR spectroscopic analysis of 1 shows one hypersilyl singlet resonance in the 1H NMR spectrum at 0.38 ppm as well as resonances at 2.58, 3.57 and 3.61 ppm from the two [K(2,2,2-crypt)]+ counter-ions. 13C{1H} and 1H/29Si HMBC NMR spectra are in agreement with retention of the D3d geometry in solution, the latter exhibiting two 29Si NMR resonances with two cross-peaks. This is as expected from the symmetry observed in the solid state. Owing to decomposition via fragmentation in solution, a small amount (∼3%) of the previously reported [K(2,2,2-crypt)][Ge9(Hyp)3] is also present (see ESI†).
In an attempt to isolate a mixed sandwich complex, we turned our attention to the reaction of Ni(Cp)2 or (Cp)Ni(PPh3)Cl with K[Ge9(Hyp)3], however both reactions resulted in vertex substitution, whereby a germanium atom of the cluster cage is replaced by a (Cp)Ni fragment, producing the neutral cluster (Cp)Ni[Ge8(Hyp)3] (2;Scheme 2). Vertex substitution has been previously observed for unsubstituted [E9]4− (E = Ge, Sn) clusters,17–19 performed in highly polar solvents under reducing conditions, however such transformations are unprecedented for functionalized precursors such as [Ge9(Hyp)3]−.
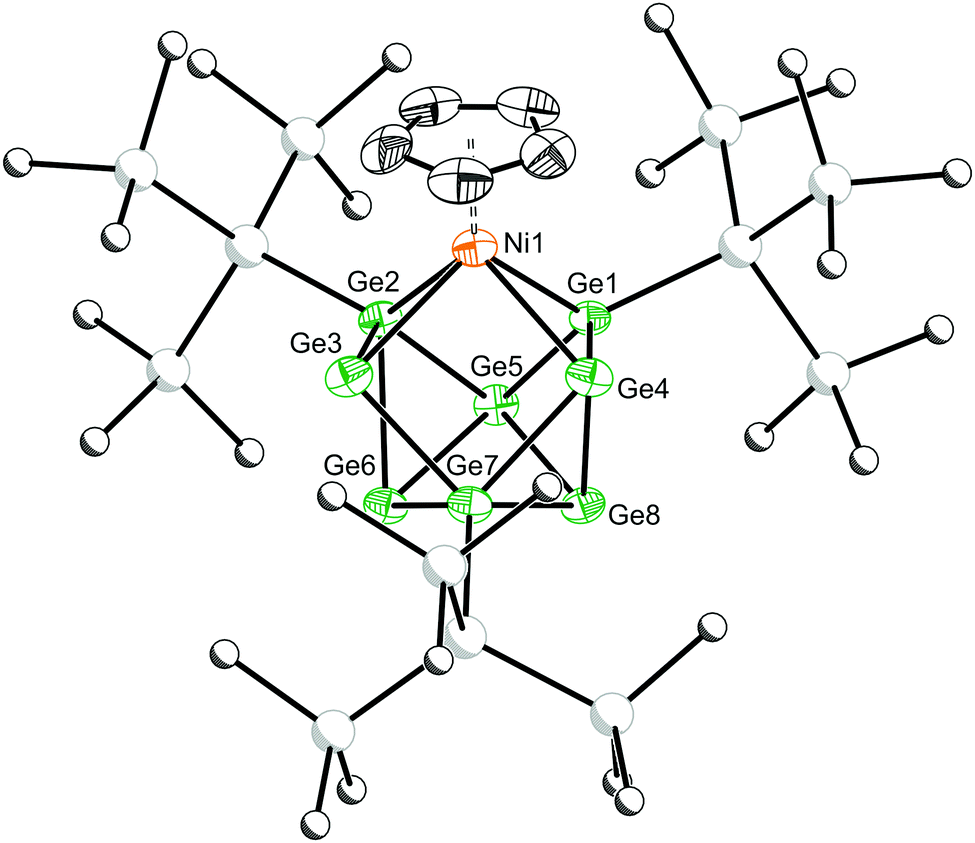 | ||
| Fig. 2 Molecular structure of 2·1.5tol. Anisotropic displacement ellipsoids are set at 50% probability. Hydrogen atoms and solvent of crystallisation have been omitted for clarity. Carbon and silicon atoms are pictured as spheres of arbitrary radii. Selected bond distances [Å]: Ni1–Ge1:2.400(2); Ni1–Ge2:2.404(2); Ni1–Ge3:2.591(2); Ni1–Ge4:2.580(2). | ||
Emerald green crystals of 2 suitable for single crystal X-ray crystallography were grown from a concentrated solution of toluene (Fig. 2). The cluster adopts a Cs-symmetric distorted mono-capped square-antiprismatic geometry, with the Ni(Cp) moiety occupying a basal position. This cluster can be described as a nido-deltahedron with 22 electrons available for cluster bonding.24 Structurally, this cluster is closely related to [(Cp)TiSn8]3− and [(CO)3FeGe8]3−.18,19
The NMR spectroscopic data are in agreement with the solid state structure, exhibiting two 1H NMR resonances for the hypersilyl environments at 0.49 and 0.60 ppm in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, in addition to a single resonance for the C5H5 ligand at 5.18 ppm. In addition to this, 13C{1H} and 1H/29Si HMBC NMR supply further evidence for the two inequivalent hypersilyl environments, with four cross peaks observed in the HMBC spectrum.
:1 ratio, in addition to a single resonance for the C5H5 ligand at 5.18 ppm. In addition to this, 13C{1H} and 1H/29Si HMBC NMR supply further evidence for the two inequivalent hypersilyl environments, with four cross peaks observed in the HMBC spectrum.
The structural relationship and contrasting electron counts between 2 and [(CO)3FeGe8]3− (22 and 21 clusters bonding electrons, respectively), point to the possibility of facile redox processes. We were intrigued to explore the electrochemical behaviour of these clusters through a series of cyclic voltammetry (CV) studies.
The dianionic sandwich complex 1 features a reversible oxidation at E01 = −1.56 V (Fig. 3), which is followed by a second, quasi-reversible oxidation event at E02 = −1.20 V (all potentials given relative to the ferrocene/ferrocenium redox couple; Fc0/+ = 0). This is followed by a third oxidation (E3pa = −0.85 V vs. Fc0/+) which is irreversible at all scan rates (ν = 0.05–1 mV s−1). To date, this is only the second observation of reversible redox behaviour for solutions of Zintl clusters.25 However, attempts to chemically access these oxidized clusters by, for example, oxidation with cobaltocenium hexafluorophosphate were unsuccessful. This leads us to believe that while oxidized clusters may be kinetically accessible, they cannot be accessed because of decomposition.
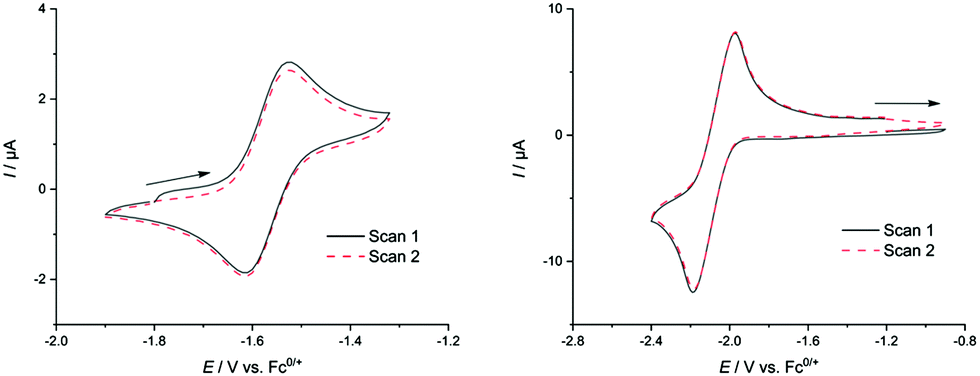 | ||
| Fig. 3 Cyclic voltammogram of complexes 1 (left; 100 mV s−1; first oxidative event; 1.0 mM, 0.2 M [NBu4][PF6], THF, room temperature) and 2 (right; 100 mV s−1; first reductive event; 1.0 mM, 0.1 M [NBu4][PF6], THF, room temperature). Arrows indicated starting points of scans and their direction. | ||
Electrochemical analysis of 2 features a reversible reductive event at E01 = −2.07 V. This is followed by a second reduction at E2pc = −2.54 V, which is irreversible at all scan rates (ν = 0.05–1 mV s−1) and is accompanied by two additional irreversible features in the reverse scan (Epa = −2.38, −1.21 V) indicating some form of cluster degradation/rearrangement on over-reduction. Attempts to access this compound via reduction with a Na/Hg amalgam gave rise to a complex 1H NMR spectrum, indicative of excessive cluster decomposition. As with 1, it would appear that while these reversible reductive redox events are observable in the cyclic voltammetry measurements, chemical isolation of these compounds is challenging. Interestingly, and despite the precedent for a closely related oxidized cluster, [(CO)3Fe(Ge8)]3−, no oxidation events were observed in the CV scans.
We have shown that two distinct reactivity pathways are accessible for the functionalised Zintl cluster [Ge9(Hyp)3]− on reaction with nickel reagents. In the case of the nickel(0) precursor Ni(COD)2 simple ligand displacement gives rise to the sandwich type compound [Ni{Ge9(Hyp)3}2]2−, and cluster expansion. By contrast, reactions with the nickel(II) reagents Ni(Cp)2 or (Cp)Ni(PPh3)Cl were found to give rise to vertex-substitution reactions, whereby a germanium atom of the [Ge9(Hyp)3]− cluster is replaced by a Ni(Cp) moiety. This is the first example of such a transformation involving functionalized Zintl clusters such as [Ge9(Hyp)3]−. While the reaction mechanism for such a transformation remains unknown, the high yield in which this compound can be obtained should allow for further reactivity studies.
We thank Shell Global Solutions International B. V., the EPSRC and the University of Oxford for financial support of this research (Industrial CASE studentship O. P. E. T.). The University of Oxford is also acknowledged for access to Chemical Crystallography facilities.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Y. Chen, S. L. Jheng and H. Y. Tuan, Nanoscale, 2018, 10, 11072–11078 RSC; (b) P. W. Menezes, S. Yao, R. Beltrán-Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 4640–4647 CrossRef CAS PubMed.
- (a) B. De Schutter, K. Van Stiphout, N. M. Santos, E. Bladt, J. Jordan-Sweet, S. Bals, C. Lavoie, C. M. Comrie, A. Vantomme and C. Detavernier, J. Appl. Phys., 2016, 119, 135305 CrossRef; (b) T. Grzela, G. Capellini, W. Koczorowski, M. A. Schubert, R. Czajka, N. J. Curson, I. Heidmann, T. Schmidt, J. Falta and T. Schroeder, Nanotechnology, 2015, 26, 385701 CrossRef CAS PubMed; (c) M. Sheehan, Y. Guo, G. Flynn, H. Geaney and K. M. Ryan, CrystEngComm, 2017, 19, 2072–2078 RSC; (d) M. Swain, S. Singh, D. Bhattacharya, A. Singh, R. B. Tokas, C. L. Prajapat and S. Basu, AIP Adv., 2015, 5, 077129 CrossRef; (e) C. Yan, J. M. Higgins, M. S. Faber, P. S. Lee and S. Jin, ACS Nano, 2011, 5, 5006–5014 CrossRef CAS PubMed; (f) S. Zhu and A. Nakajima, Jpn. J. Appl. Phys., Part 2, 2005, 44, L753 CrossRef CAS.
- (a) K. Kim, S. Mun, M. Jang, J. Sok and K. Park, Appl. Phys. A: Mater. Sci. Process., 2021, 127, 50 CrossRef CAS; (b) M. Noroozi, B. Hamawandi, M. S. Toprak and H. H. Radamson, ULIS 2014–2014 15thInt. Conf. Ultim. Integr. Silicon, IEEE Computer Society, 2014, 125–128.
- For review articles see: (a) B. Weinert, S. Mitzinger and S. Dehnen, Chem. – Eur. J., 2018, 24, 8470–8490 CrossRef CAS PubMed; (b) R. J. Wilson, B. Weinert and S. Dehnen, Dalton Trans., 2018, 47, 14861–14869 RSC; (c) B. Weinert and S. Dehnen, Struct. Bonding, 2017, 174, 99–134 CrossRef; (d) N. Korber, Angew. Chem., Int. Ed., 2009, 48, 3216–3217 CrossRef CAS PubMed; (e) S. C. Sevov and J. M. Goicoechea, Organometallics, 2006, 25, 5678–5692 CrossRef CAS.
- For a recent review of intermetalloid clusters see: K. Mayer, J. Weßing, T. F. Fässler and R. A. Fischer, Angew. Chem., Int. Ed., 2018, 57, 14372–14393 CrossRef CAS PubMed.
- J. M. Goicoechea and S. C. Sevov, J. Am. Chem. Soc., 2006, 128, 4155–4161 CrossRef CAS PubMed.
- J. M. Goicoechea and S. C. Sevov, Angew. Chem., Int. Ed., 2005, 117, 4094–4096 CrossRef.
- C. Liu, L. J. Li, Q. J. Pan and Z. M. Sun, Chem. Commun., 2017, 53, 6315–6318 RSC.
- E. N. Esenturk, J. Fettinger and B. Eichhorn, Polyhedron, 2006, 25, 521–529 CrossRef CAS.
- (a) C. Zhang, H. W. T. Morgan, Z. C. Wang, C. Liu, Z. M. Sun and J. E. McGrady, Dalton Trans., 2019, 48, 15888–15895 RSC; (b) S. Mitzinger, L. Broeckaert, W. Massa, F. Weigend and S. Dehnen, Nat. Commun., 2016, 7, 1–10 Search PubMed; (c) J. Q. Wang, S. Stegmaier, B. Wahl and T. F. Fässler, Chem. – Eur. J., 2010, 16, 1793–1798 CrossRef CAS PubMed.
- (a) F. Li and S. C. Sevov, Inorg. Chem., 2012, 51, 2706–2708 CrossRef CAS PubMed; (b) A. Schnepf, Angew. Chem., Int. Ed., 2003, 42, 2624–2625 CrossRef CAS PubMed.
- For examples of η1-[Ge9R3]− clusters see: (a) F. S. Geitner, W. Klein, O. Storcheva, T. D. Tilley and T. F. Fässler, Inorg. Chem., 2019, 58, 13293–13298 CrossRef CAS PubMed; (b) N. C. Michenfelder, C. Gienger, A. Schnepf and A. N. Unterreiner, Dalton Trans., 2019, 48, 15577–15582 RSC.
- For examples of η3-[Ge9R3]− clusters see: (a) L. J. Schiegerl, M. Melaimi, D. R. Tolentino, W. Klein, G. Bertrand and T. F. Fässler, Inorg. Chem., 2019, 58, 3256–3264 CrossRef CAS PubMed; (b) O. Kysliak, D. D. Nguyen, A. Z. Clayborne and A. Schnepf, Inorg. Chem., 2018, 57, 12603–12609 CrossRef CAS PubMed; (c) F. S. Geitner, M. A. Giebel, A. Pöthig and T. F. Fässler, Molecules, 2017, 22, 1204 CrossRef PubMed; (d) F. S. Geitner and T. F. Fässler, Eur. J. Inorg. Chem., 2016, 2688–2691 CrossRef CAS; (e) O. Kysliak, C. Schrenk and A. Schnepf, Chem. – Eur. J., 2016, 22, 18787–18793 CrossRef CAS PubMed; (f) K. Mayer, L. J. Schiegerl and T. F. Fässler, Chem. – Eur. J., 2016, 22, 18794–18800 CrossRef CAS PubMed; (g) F. Li and S. C. Sevov, Inorg. Chem., 2015, 54, 8121–8125 CrossRef CAS PubMed; (h) C. Schenk, F. Henke, G. Santiso-Quiñones, I. Krossing and A. Schnepf, Dalton Trans., 2008, 4436–4441 RSC; (i) F. Henke, C. Schenk and A. Schnepf, Dalton Trans., 2009, 9141–9145 RSC; (j) C. Schenk and A. Schnepf, Angew. Chem., Int. Ed., 2007, 46, 5314–5316 CrossRef CAS PubMed.
- For examples of η5-[Ge9R3]− clusters see: (a) S. Frischhut, F. Kaiser, W. Klein, M. Drees, F. E. Kühn and T. F. Fässler, Organometallics, 2018, 37, 4560–4567 CrossRef CAS; (b) F. Li, A. Muñoz-Castro and S. C. Sevov, Angew. Chem., Int. Ed., 2016, 55, 8630–8633 CrossRef CAS PubMed; (c) F. Henke, C. Schenk and A. Schnepf, Dalton Trans., 2011, 40, 6704–6710 RSC; (d) C. Schenk and A. Schnepf, Chem. Commun., 2009, 3208–3210 RSC.
- O. P. E. Townrow, C. Chung, S. A. Macgregor, A. S. Weller and J. M. Goicoechea, J. Am. Chem. Soc., 2020, 142, 18330–18335 CrossRef CAS PubMed.
- D. R. Gardner, J. C. Fettinger and B. W. Eichhorn, Angew. Chem., Int. Ed. Engl., 1996, 35, 2852–2854 CrossRef CAS.
- For a recent example of vertex substitution reactions in Zintl cluster chemistry see: A. M. Li, Y. Wang, P. Y. Zavalij, F. Chen, A. Muñoz-Castro and B. W. Eichhorn, Chem. Commun., 2020, 56, 10859–10862 RSC.
- C. B. Benda, M. Waibel and T. F. Fässler, Angew. Chem., Int. Ed., 2015, 54, 522–526 CAS.
- B. Zhou and J. M. Goicoechea, Chem. – Eur. J., 2010, 16, 11145–11150 CrossRef CAS PubMed.
- E. N. Esenturk, A. J. C. Fettinger and B. W. Eichhorn, J. Am. Chem. Soc., 2006, 128, 12–13 CrossRef CAS PubMed.
- (a) J. Bachmann, A. Zolotaryov, O. Albrecht, S. Goetze, A. Berger, D. Hesse, D. Novikov and K. Nielsch, Chem. Vap. Deposition, 2011, 17, 177–180 CrossRef CAS; (b) S.-D. Hwang, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct. –Process., Meas., Phenom., 1996, 14, 2957 CrossRef CAS; (c) M. V. Kharlamova, M. Sauer, T. Saito, Y. Sato, K. Suenaga, T. Pichler and H. Shiozawa, Nanoscale, 2015, 7, 1383–1391 RSC; (d) J. A. Singh, N. F. W. Thissen, W. H. Kim, H. Johnson, W. M. M. Kessels, A. A. Bol, S. F. Bent and A. J. M. MacKus, Chem. Mater., 2018, 30, 663–670 CrossRef CAS PubMed.
- (a) J. S. Bradley, B. Tesche, W. Busser, M. Maase and M. T. Reetz, J. Am. Chem. Soc., 2000, 122, 4631–4636 CrossRef CAS; (b) N. J. S. Costa, R. F. Jardim, S. H. Masunaga, D. Zanchet, R. Landers and L. M. Rossi, ACS Catal., 2012, 2, 925–929 CrossRef CAS; (c) T. O. Ely, C. Amiens, B. Chaudret, E. Snoeck, M. Verelst, M. Respaud and J. M. Broto, Chem. Mater., 1999, 11, 526–529 CrossRef CAS.
- O. Kysliak and A. Schnepf, Dalton Trans., 2016, 45, 2404–2408 RSC.
- T. A. Albright, J. K. Burdett and M. H. Whangbo, Orbital Interactions in Chemistry, 2nd edn, 2013 Search PubMed.
- J. M. Goicoechea and S. Sevov, Inorg. Chem., 2005, 44, 2654–2658 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data, spectra and computational methods. CCDC 2087498 and 2087499. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc02912f |
| ‡ See ESI for all experimental details. |
| This journal is © The Royal Society of Chemistry 2021 |