
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct formation of 4,5-disubstituted carbazoles via regioselective dilithiation†
Ian A.
Pocock
 a,
Alya M.
Alotaibi
ab,
Kesar
Jagdev
a,
Connor
Prior
a,
Gregory R.
Burgess
a,
Louise
Male
a and
Richard S.
Grainger
*a
a,
Alya M.
Alotaibi
ab,
Kesar
Jagdev
a,
Connor
Prior
a,
Gregory R.
Burgess
a,
Louise
Male
a and
Richard S.
Grainger
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: r.s.grainger@bham.ac.uk
bPrince Sattam bin Abdulaziz University, Saudi Arabia
First published on 18th June 2021
Abstract
Carbazoles are widely exploited for their interesting photophysical and electronic properties, however bay (4,5-) functionalization is challenging, and previously inaccessible through carbazole C–H activation. We report a simple methodology which introduces a range of versatile 4,5-functionality, enabling the wider investigation of ring annulation and close proximity effects on carbazole properties.
Carbazoles represent a privileged structure within chemistry: their advantageous photophysical and electronic properties have led to multiple applications in organic materials,1 sensing,2 and as organic photocatalysts.3 Carbazoles are also found in a large number of naturally-occurring alkaloids4 and other biologically active molecules,5 and hence the development of methods for their preparation remains an important area of research.6
Groups at the 4- and 5-positions, or bay region, of carbazole are in close proximity, but systematic investigations into such peri-like interactions are limited by the lack of effective preparatory methods and general synthetic building blocks. Thus the relatively few examples of 4,5-disubstituted carbazoles in the literature are prepared from non-carbazole precursors through benzannulation of indoles or pyrroles,6b or through formation of the central nitrogen heterocycle from biaryls or diarylamines.6c The influence of close proximity, peri-like interactions on the structure, reactivity and electronic properties of 4,5-disubstituted carbazoles has been noted in a few cases,7,8 and carbazoles with additional ring fusion at the 4,5-positions have been investigated in a variety of contexts, but again not synthesized by annulation of carbazole.9
Aromatic C–H functionalization represents the most direct method for the preparation of substituted carbazoles, however the ease of C-substitution is highly positional dependent (Scheme 1). Functionalization at the 3,6-positions (through electrophilic aromatic substitution10) and at the 1,8-positions (through directed lithiation11 or metal-catalysed C–H activation12) are well established. Other substitution patterns are more difficult to access: 2,7-functionalization is rare,13 and there is only one method for C–H activation at C-4, through the coupling of N-pyrimidinyl carbazoles with tertiary α-bromoesters under Ru catalysis.14
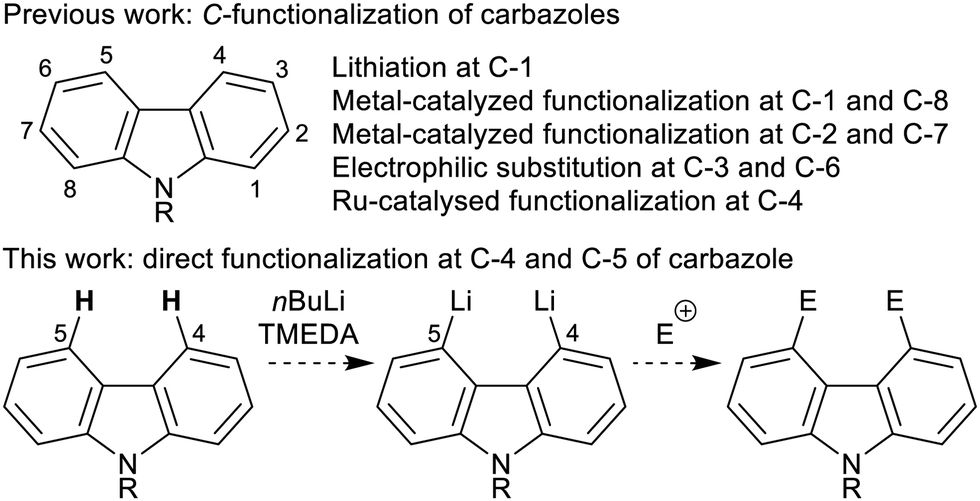 | ||
| Scheme 1 Considerations in carbazole functionalization. | ||
The combination of nBuLi and N,N,N′,N′-tetramethylethylenediamine (TMEDA) can be used to regioselectively dilithiate a select range of aromatic systems through directed deprotonation.15 Given the known effect of placing a triisopropylsilyl (TIPS) group on an indole nitrogen to block ortho-lithiation,16 we hypothesized that the combination of a bulky silyl protecting group on the carbazole nitrogen (to prevent C-1/C-8-lithiation) and the use of nBuLi–TMEDA (to dilithiate at C-4 and C-5)17 would offer a simple and convenient approach to 4,5-difunctionalized carbazoles upon electrophilic trapping (Scheme 1). Based on this hypothesis, we herein report the first method for the direct functionalization at both the 4- and 5-positions of carbazole.
9H-Carbazole (1b) and silyl-substituted carbazoles 1a (R = TIPS),211c (R = TES) and 1d (R = TBS) were treated with nBuLi under various conditions, followed by quenching with D2O, to determine the extent and position of lithiation, and the functional group compatibility of the N-silyl group (Table 1). Subjecting N-TIPS carbazole 1a to standard nBuLi–TMEDA conditions15a,c gave 70% deuterium incorporation at the 4,5-positions, as estimated by 1H NMR (Table 1, entry 1),18 thus establishing the feasibility of the approach. Extending the reaction time (entries 2 and 3) and increasing the reaction temperature (entries 4 and 5) gave increased levels of deuterium incorporation with the same C-4,5 regioselectivity; however evaporation of hexanes proved troublesome, thus favouring longer reaction times as the more practical conditions (entry 2). A screen of N-substituents showed that the free 9H-carbazole (1b) gave deuteration preferentially at C-1 (entry 6), complementing prior literature in the absence of TMEDA.11a Smaller N-silyl substituents resulted in extensive desilylation (entries 7 and 8). Finally, exploring the role of the ligand, attempting the lithiation in the absence of TMEDA gave no deuterium incorporation (entry 9), while using PMDTA, a known ligand for remote lithiation,19 resulted in decomposition of 1a (entry 10).
The conditions from Table 1, entry 2 were screened with a range of different electrophiles to give 4,5-disubstituted N-TIPS carbazoles directly from N-TIPS carbazole 1a (Fig. 1). Alkylation with iodomethane gave a mixture of starting material, mono- and dimethylated products from which the major component, 4,5-dimethylcarbazole 2a, could be separated in 38% yield. However the larger allyl bromide and benzyl bromide electrophiles failed to react.20 Results of trapping with other carbon electrophiles also highlighted the difficulty in incorporating two groups in close proximity in the carbazole bay region. Although reaction with benzophenone was unsuccessful, use of paraformaldehyde gave separable bis-alcohol 3a21 in 43% yield, along with 23% of the monoalcohol. Reaction with CO2 gave anhydride 4a in a pleasing 63% yield. Trapping with hexafluorobenzene allowed for annulation to the 4H-naphtho[1,2,3,4-def]carbazole 5a in 34% yield.21
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 1 (R) | Ligand | Time (h) | Temp. (°C) | % deuterationa | ||
| C1/8 | C2–6 | C4/5 | |||||
| a Determined by relative integration of aromatic hydrogen signals in the 1H NMR. | |||||||
| 1 | 1a (TIPS) | TMEDA | 3 | 60 | — | — | 70 |
| 2 | 1a (TIPS) | TMEDA | 6 | 60 | — | — | 83 |
| 3 | 1a (TIPS) | TMEDA | 9 | 60 | — | — | 75 |
| 4 | 1a (TIPS) | TMEDA | 3 | 68 | — | — | 83 |
| 5 | 1a (TIPS) | TMEDA | 6 | 68 | — | — | 79 |
| 6 | 1b (H) | TMEDA | 6 | 60 | 57 | — | 12 |
| 7 | 1c (TES) | TMEDA | 6 | 60 | 100% desilylation | ||
| 8 | 1d (TBS) | TMEDA | 6 | 60 | 88% desilylation | ||
| 9 | 1a (TIPS) | — | 6 | 60 | — | — | — |
| 10 | 1a (TIPS) | PMDTA | 6 | 60 | Decomposition | ||
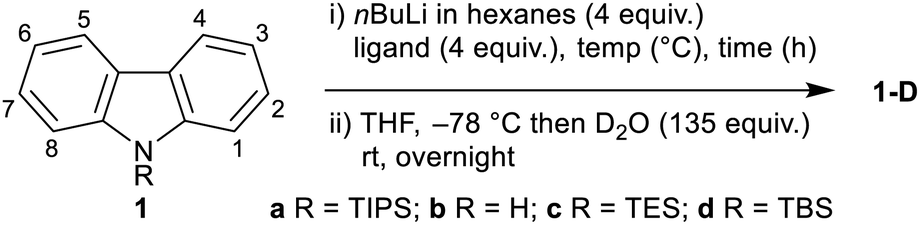
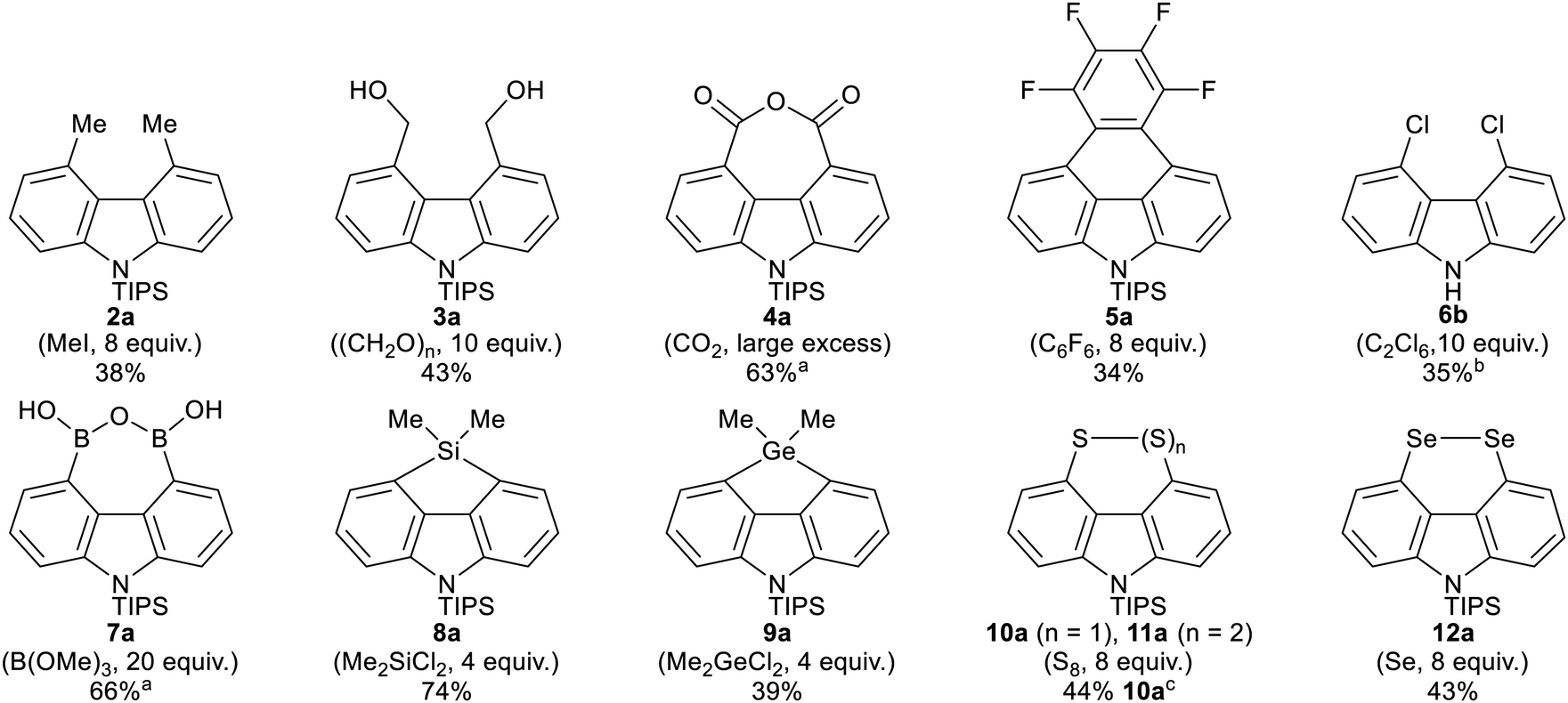 | ||
Fig. 1 4,5-Dilithiation and electrophilic trapping of N-TIPS carbazole 1a. Reaction conditions: (i) nBuLi (4 equiv.), TMEDA (4 equiv.), 60 °C, 6 hours, (ii) THF, −78 °C then (electrophile, equiv.). % yields refer to isolated yields after purification by column chromatography unless otherwise stated. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated yield after purification by trituration with n-hexane. bIsolated yield over two steps after removal of TIPS group with TBAF. cYield of 10a after NaBH4 reduction of mixture of 10a and 11a. Isolated yield after purification by trituration with n-hexane. bIsolated yield over two steps after removal of TIPS group with TBAF. cYield of 10a after NaBH4 reduction of mixture of 10a and 11a. | ||
Reaction with heteroatom electrophiles was also investigated. Chlorination with C2Cl6 gave a mixture of monochloride and dichloride, which were separable after removal of the TIPS group from the carbazole nitrogen using TBAF, with dichloride 6b21 isolated in 35% yield, along with 13% of the monochloride. Attempted incorporation of larger bromine or iodine were, however, very low yielding, again highlighted the difficulty in incorporating two groups in close proximity in the carbazole bay region.20 More successful was where the electrophile was particularly small, or where a ring could be formed in situ, relieving potential peri-like steric and electronic repulsions in the hindered carbazole bay region. Reaction with B(OMe)3 gave boronic anhydride 7a in 66% yield after acidic work-up.22 Reaction with dimethyldichlorosilane gave silafluorene 8a in 74% yield.21 Germafluorene 9a was similarly formed in 39% yield.
Our interest in peri-substituted naphthalene disulfides and diselenides led us to investigate incorporation of sulfur and selenium at C-4 and C-5.23 Reaction with elemental sulfur gave disulfide 10a in 54% yield on a 3.1 mmol scale. On larger scale (6.2 mmol 1a), mixtures of disulfide 10a and trisulfide 11a were obtained, which could be converged to 10a in 44% overall yield by treatment with NaBH4. Reaction with elemental selenium gave diselenide 12a in 43% yield without evidence of any triselenide formation. X-Ray analyses of disulfide 10b and diselenide 12b, obtained after removal of the N-TIPS groups with TBAF, are reported in the ESI.†21
The functionality embedded in these novel 4,5-disubstituted carbazoles can be further manipulated, increasing the range of carbazoles accessible from this methodology (Scheme 2). Regioselective electrophilic aromatic substitution of the fused carbazole–silafluorene 8a provided the corresponding 3,6-disubstituted carbazoles 13a–16a. Silafluorenes have broad application in materials and synthesis,24 and the preparation of dihalides 13a–15a in good yields opens up the investigation of these novel heteroatom-bridged heterofluorenes25 in further cross-coupling reactions. Oxidation of 8a under Dudley's conditions gave desymmetrized carbazole monoalcohol 17a.26N-Desilylation of 17a with TBAF gave 4-hydroxycarbazole (17b), a known starting point for natural product syntheses.27 Complementary to the mono-oxidation of 8a, the diol 18a was the major product formed in the oxidation of boronic anhydride 7a using NMO.28 The potential of anhydride 4a was demonstrated through reaction with hydrazine hydrate to give the N-iminoimide 19a in 30% yield (unoptimized). Dehydration of diol 3a to the carbazole-annulated cyclic ether 20a21 was achieved using H3PO4.29 Oxidative insertion of Fe3(CO)12 into the disulfide 10a gave the Fe2(CO)6 complex 21a, of interest as a structural mimic of the active site of the enzyme [FeFe] hydrogenase.23d,30
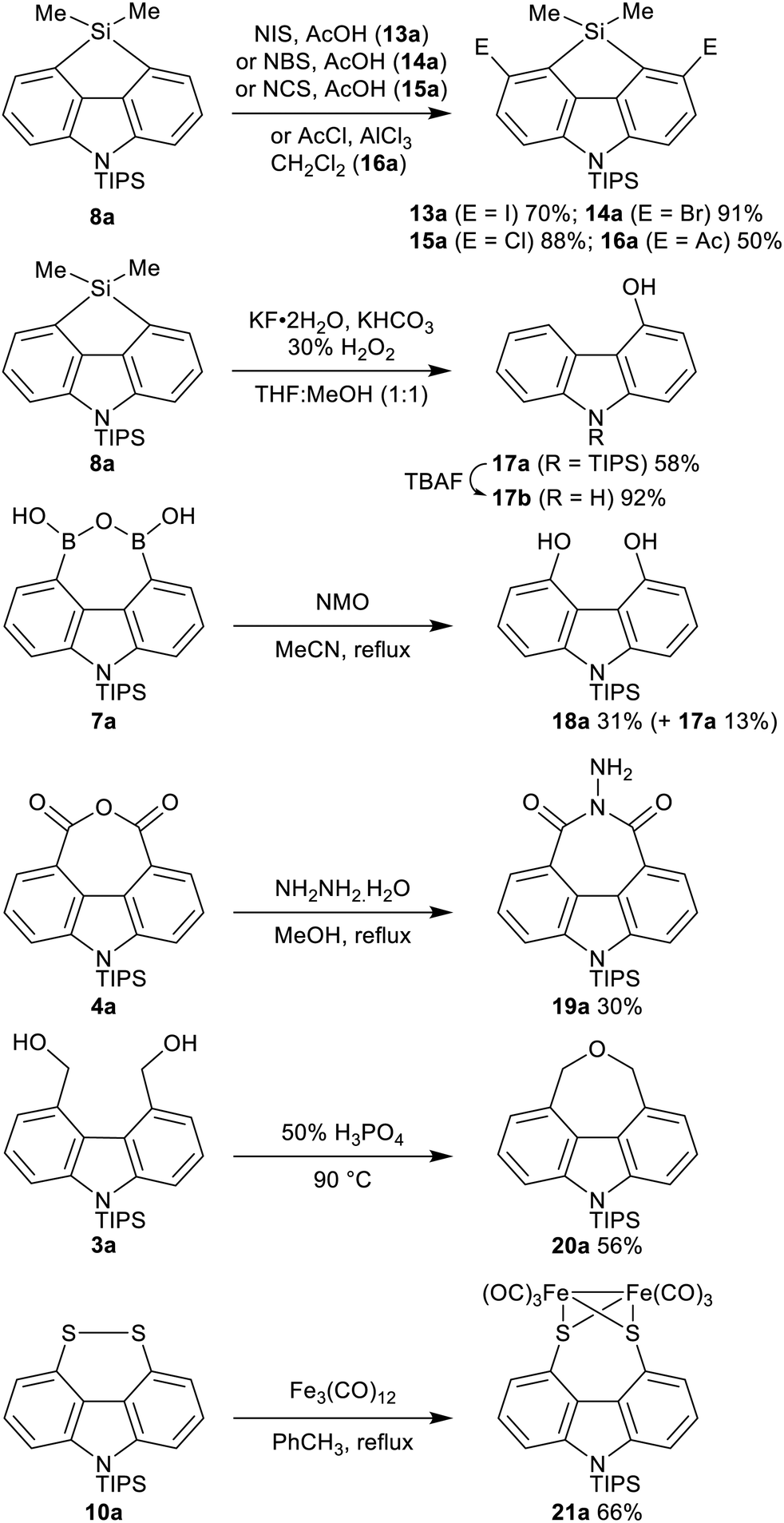 | ||
| Scheme 2 Selective transformations of 4,5-disubstituted carbazoles. | ||
In conclusion, the first methodology for the direct functionalization of the sterically hindered bay positions of carbazoles is reported, through the simple combination of a bulky TIPS group on the carbazole nitrogen and the use of nBuLi–TMEDA for deprotonation and directed lithiation. With 4,5-substitution being relatively unrepresented in the carbazole literature, and substitution in this bay region effecting, for example, the planarity of the carbazole,8,31 this methodology opens up new opportunities for more systematic investigations and future applications in synthesis, catalysis, organic materials and biology.
We thank the Leverhulme Trust (RPG-2017-260), Prince Sattam bin Abdulaziz University, Saudi Arabia, and EPSRC (EP/M508202/1, EP/N509590/1) for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Recent reviews: (a) D. Devadiga, M. Selvakumar, P. Shetty, M. S. Santosh, R. S. Chandrabose and S. Karazhanov, Int. J. Energy Res., 2021, 45, 6584–6643 CrossRef CAS; (b) F.-M. Xie, J.-X. Zhou, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2020, 8, 9476–9494 RSC; (c) F. Bekkar, F. Bettahar, I. Moreno, R. Meghabar, M. Hamadouche, E. Hernáez, J. L. Vilas-Vilela and L. Ruiz-Rubio, Polymers, 2020, 12, 2227 CrossRef CAS PubMed; (d) G. Krucaite and S. Grigalevicius, Synth. Met., 2019, 247, 90–108 CrossRef CAS; (e) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (f) B. Wex and B. R. Kaafarani, J. Mater. Chem. C, 2017, 5, 8622–8653 RSC.
- Recent reviews: (a) J. Yin, Y. Ma, G. Li, M. Peng and W. Lin, Coord. Chem. Rev., 2020, 412, 213257 CrossRef CAS; (b) I. Gupta and P. E. Kesavan, Front. Chem., 2019, 7, 841 CrossRef CAS PubMed.
- (a) T. Yabuta, M. Hayashi and R. Matsubara, J. Org. Chem., 2021, 86, 2545–2555 CrossRef CAS PubMed; (b) J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 13719–13725 CrossRef CAS PubMed; (c) E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed; (d) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed.
- Reviews: (a) T. Janosik, A. Rannug, U. Rannug, N. Wahlström, J. Slätt and J. Bergman, Chem. Rev., 2018, 118, 9058–9128 CrossRef CAS PubMed; (b) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193–3328 CrossRef CAS PubMed.
- Recent reviews: (a) A. Caruso, J. Ceramella, D. Iacopetta, C. Saturnino, M. V. Mauro, R. Bruno, S. Aquaro and M. S. Sinicropi, Molecules, 2019, 24, 1912 CrossRef CAS PubMed; (b) S. Issa, A. Prandina, N. Bedel, P. Rongved, S. Yous, M. L. Borgne and Z. Bouaziz, J. Enzyme Inhib. Med. Chem., 2019, 34, 1321–1346 CrossRef CAS PubMed; (c) S. Sellamuthu, G. Gutti, D. Kumar and S. K. Singh, Mini-Rev. Org. Chem., 2018, 15, 498–507 CrossRef CAS.
- Reviews: (a) S. N. Georgiades and P. G. Nicolaou, Adv. Heterocycl. Chem., 2019, 129, 1–88 CrossRef CAS; (b) A. Banerjee, S. Kundu, A. Bhattacharyya, S. Sahu and M. S. Maji, Org. Chem. Front., 2021, 8, 2710–2771 RSC; (c) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466–478 CrossRef CAS.
- Y. Tsunashima and M. Kuroki, J. Heterocycl. Chem., 1981, 18, 715–718 CrossRef CAS.
- T. Gensch, M. Rönnefahrt, R. Czerwonka, A. Jäger, O. Kataeva, I. Bauer and H.-J. Knölker, Chem. – Eur. J., 2012, 18, 770–776 CrossRef CAS PubMed.
- (a) Z. M. Geng, K. Shibasakia and M. Kijima, Synth. Met., 2016, 213, 57–64 CrossRef CAS; (b) X.-D. Xiong, C.-L. Deng, X.-S. Peng, Q. Miao and H. N. C. Wong, Org. Lett., 2014, 16, 3252–3255 CrossRef CAS PubMed; (c) G. Mitchell and C. W. Rees, J. Chem. Soc., Perkin Trans. 1, 1987, 403–412 RSC.
- For a recent example see: X.-T. Wu, E.-K. Xiao, F. Ma, J. Yin, J. Wang, P. Chen and Y.-J. Jiang, J. Org. Chem., 2021, 86, 6734–6743 CrossRef CAS PubMed.
- (a) A. Hallberg and A. R. Martin, J. Heterocycl. Chem., 1984, 21, 837–840 CrossRef CAS; (b) A. R. Katritzky, G. W. Rewcastle and L. M. Vazquez de Miguel, J. Org. Chem., 1988, 53, 794–799 CrossRef CAS.
- Z.-C. Qi, Q.-X. Lou, Y. Niu and S.-D. Yang, Chem. Commun., 2021, 57, 2021–2024 RSC and references therein.
- (a) Y. Feng, D. Holte, J. Zoller, S. Umemiya, L. R. Simke and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 10160–10163 CrossRef CAS PubMed; (b) G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R.-Y. Tang, M. Movassaghi and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807–10813 CrossRef CAS PubMed; (c) For direct 1,2-substitution see: D. Wang, M. Li, X. Chen, M. Wang, Y. Liang, Y. Zhao, K. N. Houk and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 7066–7071 CrossRef CAS PubMed.
- J. A. Leitch, C. J. Heron, J. McKnight, G. Kociok-Köhn, Y. Bhonoah and C. G. Frost, Chem. Commun., 2017, 53, 13039–13042 RSC.
- (a) A. J. Ashe III, J. W. Kampf and P. M. Savla, Heteroat. Chem., 1994, 5, 113–119 CrossRef; (b) X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535–538 CrossRef CAS PubMed; (c) V. B. Bonifácio, J. Morgado and U. Scherf, Synlett, 2010, 1333–1336 CrossRef; (d) S. Cossu, G. Delogu, D. Fabbri and P. Maglioli, Org. Prep. Proced. Int., 1991, 23, 455–457 CrossRef CAS; (e) I. A. Kashulin and I. E. Nifant’ev, J. Org. Chem., 2004, 69, 5476–5479 CrossRef CAS PubMed.
- M. Matsuzono, T. Fukuda and M. Iwao, Tetrahedron Lett., 2001, 42, 7621–7623 CrossRef CAS.
- For rationales of bay-region and peri-dilithiation regioselectivity using nBuLi–TMEDA in related polyaromatics and the potential for bridging dilithiated species, see: (a) W. Bauer, T. Clark and P. V. R. Schleyer, J. Am. Chem. Soc., 1987, 109, 970–977 CrossRef CAS; (b) A. J. Ashe, III, J. W. Kampf and P. M. Savla, J. Org. Chem., 1990, 55, 5558–5559 CrossRef and references therein.
- G. Rouquet, D. C. Blakemore and S. V. Ley, Chem. Commun., 2014, 50, 8908–8911 RSC.
- A. B. Bellan and P. Knochel, Angew. Chem., Int. Ed., 2019, 58, 1838–1841 CrossRef CAS PubMed and references therein.
- Additional electrophiles which were unsuccessful in an initial trapping screen (no reaction or complex mixtures leading to poor yields) are reported in the ESI†.
- CCDC 2079909–2079912 (1a, 8a, 10b and 12b respectively) and CCDC 2080733–2080736 (3a, 5a, 6b and 20a respectively) contain the supplementary crystallographic data for this paper†.
- (a) R. L. Letsinger, J. M. Smith, J. Gilpin and D. B. MacLean, J. Org. Chem., 1965, 30, 807–812 CrossRef CAS; (b) A. Das, A. Hübner, M. Weber, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2011, 47, 11339–11341 RSC.
- (a) R. S. Grainger, B. Patel and B. M. Kariuki, Angew. Chem., Int. Ed., 2009, 48, 4832–4835 CrossRef CAS PubMed; (b) R. S. Grainger, B. Patel, B. M. Kariuki, L. Male and N. Spencer, J. Am. Chem. Soc., 2011, 133, 5843–5852 CrossRef CAS PubMed; (c) B. Patel, J. Carlisle, S. E. Bottle, G. R. Hanson, B. M. Kariuki, L. Male, J. C. McMurtrie, N. Spencer and R. S. Grainger, Org. Biomol. Chem., 2011, 9, 2336–2344 RSC; (d) C. Figliola, L. Male, S. L. Horswell and R. S. Grainger, Eur. J. Inorg. Chem., 2015, 3146–3156 CrossRef CAS.
- (a) For materials applications see: Y. Dong, Y. Takata, Y. Yoshigoe, K. Sekine and Y. Kuninobu, Chem. Commun., 2019, 55, 13303–13306 RSC and references therein; ; (b) For synthetic applications see: H. Ito, Y. Segawa, K. Murakami and K. Itami, J. Am. Chem. Soc., 2019, 141, 3–10 CrossRef CAS PubMed.
- H. Sasaki, I. Akioka, H. Imoto and K. Naka, Eur. J. Org. Chem., 2021, 1390–1395 CrossRef CAS.
- J. D. Sunderhaus, H. Lam and G. B. Dudley, Org. Lett., 2003, 5, 4571–4573 CrossRef CAS PubMed.
- C. Brütting, R. Hesse, A. Jäger, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Chem. – Eur. J., 2016, 22, 16897–16911 CrossRef PubMed.
- Y. Chai, M. Zou and M. Zhu, CN107089936, 2017.
- U. Azzena, S. Demartis, L. Pilo and E. Piras, Tetrahedron, 2000, 56, 8375–8382 CrossRef CAS.
- For a review see: J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC.
- The C4–C–C–C5 dihedral angle in disulfide 10b (11.4(3)°, 10.5(3)°) and diselenide 12b (13.6(4)°) can be compared with the essentially planar carbazole ring system in 1a (−0.8(5)°). See ref. 21 for crystallography data.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, additional electrophile and substrate screen, X-ray crystallography and copies of NMR spectra. CCDC 2079909–2079912 and 2080733–2080736. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc02892h |
| This journal is © The Royal Society of Chemistry 2021 |