
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Varying the hydrophobic spacer to influence multicomponent gelation†
Santanu
Panja
 ,
Bart
Dietrich
,
Adriana
Trabold
,
Agata
Zydel
,
Aleena
Qadir
and
Dave J.
Adams
*
,
Bart
Dietrich
,
Adriana
Trabold
,
Agata
Zydel
,
Aleena
Qadir
and
Dave J.
Adams
*
School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
First published on 9th July 2021
Abstract
Mixing low molecular weight gelators (LMWGs) shows promise as a means of preparing innovative materials with exciting properties. Here, we investigate the effect of increasing hydrophobic chain length on the properties of the resulting multicomponent systems which are capable of showing ambidextrous phase behaviour on pH perturbation.
Non-covalent interactions such as hydrogen bonding, aromatic stacking, electrostatic and hydrophobic interactions are the essence of most biological systems and processes.1 Incorporation of such interactions into synthetic materials can lead to various self-assembled systems; supramolecular hydrogels are one of these that have many applications.2,3 Typically, hydrogels are formed when small organic molecules self-assemble into long anisotropic fibres.4 These fibres then entangle to form a 3D-network that immobilizes the water. The intermolecular interactions essentially control the formation of such fibres, and their subsequent growth and distribution inside the matrix. A subtle change in the intermolecular interactions can significantly influence the gel properties; perturbation of non-covalent forces may even lead to destruction of gels and formation of sols.5
In spite of reasonable progress in gel chemistry, it is still a difficult task to design molecules that form gels.4 Although computational techniques have been developed to predict gelation propensities of compounds, their use is still limited.6 As an alternative approach, it is common to adapt the properties of known gelators by following different approaches: first, by varying the route of self-assembly, specifically the self-assembly kinetics, keeping the final composition of the material identical;4,7 second, by exposing a pre-formed gel to an external stimulus like heat, pH, UV-light, or ions to thereby modify its properties.;5,8 third, by varying the chemical structure of the gelator.9 Compounds with very similar chemical structures may have significantly different gel properties. Structure–property relationship studies are essential to not only understand new functional gelators, but also to adapt material properties through tuning of the noncovalent interactions.10
A fourth approach, which has been getting increasing interest in recent years, is to synthesize multicomponent gels by mixing of two or more gelator building blocks.5,11 Multicomponent gels have several advantages over single component systems. The properties of multicomponent gels can often not be predicted from either of the individual components. For example, multicomponent gels can show a multistep gel-to-gel transition, pH responsive ambidextrous phase behaviour, improved behaviour in cell, and photoelectric properties that a single building block cannot display.12–15 Moreover, selective modulation of individual components can be used to modify the gel properties.13 In short, multicomponent gels are more complex materials than single component gels with enhanced morphogenesis12,16 and tuneable material properties.14
Here, we investigate the effect of hydrophobic spacer on gel properties in a multicomponent system. Among various non-covalent forces, hydrophobicity plays a pivotal role in biology, particularly in protein folding and assembly.1,17 In synthetic hydrogels, hydrophobicity greatly influences the gel properties. Typically, gels are prepared by reducing the solubility of gelators in solution. If the molecules are more hydrophobic, wormlike micellar aggregates can be formed in the pre-gelled solution.18 We have pointed out that, during a sol-to-gel transition, the micellar aggregates can direct the gel properties.19 Furthermore, changing hydrophobicity is reported to influence gelation kinetics,20 morphology and thermal behaviour,21 mechanical properties,22 self-healing,23 and catalytic activity24 of single component systems. The hydrophobic microenvironments within the fibre network of hydrogels have also been found to enhance stability of encapsulated materials like enzymes and proteins.25
The design principle of our system involves two different compounds with a pH-responsive carboxylic acid (compound 1) and ammonium (compound 2) functionalities (Fig. 1).15 As single building blocks, only the peptide 1 exhibits gelation at acidic pH. At high pH, both the components 1 and 2 produce sols. The components show opposite responses in their solubility and self-assembly attributes in water on pH-perturbation which allows the mixed system (1 + 2) to maintain the overall hydrophobic–hydrophilic balance enabling a gel state both at acidic and basic pH (ambidextrous phase behaviour).15 At low pH, while the gel formation occurs primarily due to self-sorting of 1, at high pH gelation was driven by the co-assembly of 1 and 2 (Scheme S1, ESI†).15 We gradually increased the chain length of the ammonium component (compounds 3 and 4) and studied the effect of hydrophobic spacer on the properties of mixed gels at both acidic and basic pH (Fig. 1). We observed that, while the properties of the multicomponent gels at low pH are hardly affected, the mechanical properties of the co-assembled gels formed at high pH increase linearly with the increase in hydrophobic structural feature of the ammonium salts. The underlying molecular packing as well as microstructures of the co-assembled gels are also influenced by the nature of the ammonium salts. We further employ the urea–urease reaction to achieve a gel-to-gel transition involving these multicomponent systems. The hydrophobicity of compounds 2–4 also influences the kinetics of pH-switchable systems.
 | ||
| Fig. 1 (a) pH responsive changes in chemical structures of the compounds 1–4. The pictures of the vials represent gel/sol states formed by the corresponding species in solution. (b) Cartoon demonstrating the pH driven changes in the self-assembled network for the multicomponent systems of 1 with (2–4). | ||
Dipeptide 1 is a well-known gelator that forms a self-supporting gel in DMSO/H2O (20/80, v/v) with a pH of 4.3 (Fig. S9, ESI†).15,26 At a pH above the apparent pKa of 1 (ca. 6.4, Fig. S10, ESI†),26 due to deprotonation of the terminal carboxylic group, the solubility of the molecules increases, and no gel formation occurs (Fig. 1 and Fig. S9, ESI†). In contrast, compounds 2–4 are highly soluble in DMSO/H2O (20/80, v/v) resulting in free-flowing solutions with pH in the range 4.9–5.2 (Fig. S9, ESI†). A UV-vis study shows that solutions of 2–4 exhibit strong absorption at 265 nm along with a shoulder peak at 300 nm (Fig. S11, ESI†). They also exhibit very similar emission profiles. In all cases, a strong emission at 318 nm corresponding to the monomeric forms of 2–4 was recorded (Fig. S12, ESI†).27 pH titration data reveal that the apparent pKa of the amphiphiles gradually increases from 8.6 to 9.1 on moving from compound 2 to 4 (Fig. S10, ESI†). At high pH, deprotonation of the terminal ammonium groups generates the corresponding amines (pH 11.1–11.3 in presence of 0.01 M NaOH, Fig. 1). There was a change in the UV-vis spectra of 2–4 at high pH (Fig. S11, ESI†). The intensity of the broad band which appeared at 258 nm for the amine form of 2 progressively decreased to 3 and 4. However, no substantial change in the absorption at 300 nm was noticed. At high pH, the monomer emission of 2–4 was red shifted by 4–5 nm along with the appearance of a new band in the region 425–525 nm (Fig. S12, ESI†). The emergence of the band at 425–525 nm suggests excimer formation in all cases due to overlapping of the fluorenyl groups at high pH.27 Interestingly, the emission intensity of both the monomer and excimer peaks increases from 2 to 4. The hydrophobicity of the amphiphiles affects the solubility of the components at high pH. In presence of NaOH, while compound 2 remained in solution, compounds 3 and 4 produced precipitation (Fig. S9, ESI†). Hence, none of the amphiphiles 2–4 have gelling capability on their own either at low pH or in presence of base.
We next prepared the multicomponent gels by mixing of compounds 1 with 2–4 in absence and presence of NaOH (0.01 M). Simple mixing of 1 and 2–4 individually results in a gel in all cases with a pH of 3.1–3.3 (Fig. S13, ESI†). These gels have very similar microstructure to that of the single component gel of 1 as observed by confocal fluorescence microscopy (Fig. 2 and Fig. S14, ESI†). In all cases, spherulitic domains of fibres were formed. In the multicomponent gels, the density of the spherulitic structure increased. We previously suggested that the slight change in the microstructure of the multicomponent gels is due to the presence of organic salts (2–4)15 which influences the hydration of the peptide in water involving the Hofmeister effect28 endowing a change in the nucleation centre and subsequent growth of the fibres. The rheological moduli (the storage and loss moduli, G′ and G′′) as well as the strain bearing capacity (critical strain or gel strength, the strain at which the gel breaks is 6–8%) of the multicomponent gels were also similar to that of the hydrogel of 1 alone (Fig. S15 and S16, ESI†). These results show that for the multicomponent gels of 1 with 2–4 at low pH, the gel matrix was formed by self-sorting. 1 forms the gel network and compounds 2–4 behave as a non-gelling additive and hardly perturb the gel properties (Scheme S1, ESI†).15
 | ||
| Fig. 2 Confocal fluorescence microscopy images (scale bars represent 20 μm) of the multicomponent gel of (1 + 2) for (a and d), (1 + 3) for (b and e) and (1 + 4) for (c and f) at pH 3.1–3.3 (a–c) and pH 10.2–10.4 (d–f). In all cases, initial concentrations of 1–4 are 2 mg mL−1, concentration of NaOH is 0.01 M, solvent is DMSO/H2O (20/80, v/v). | ||
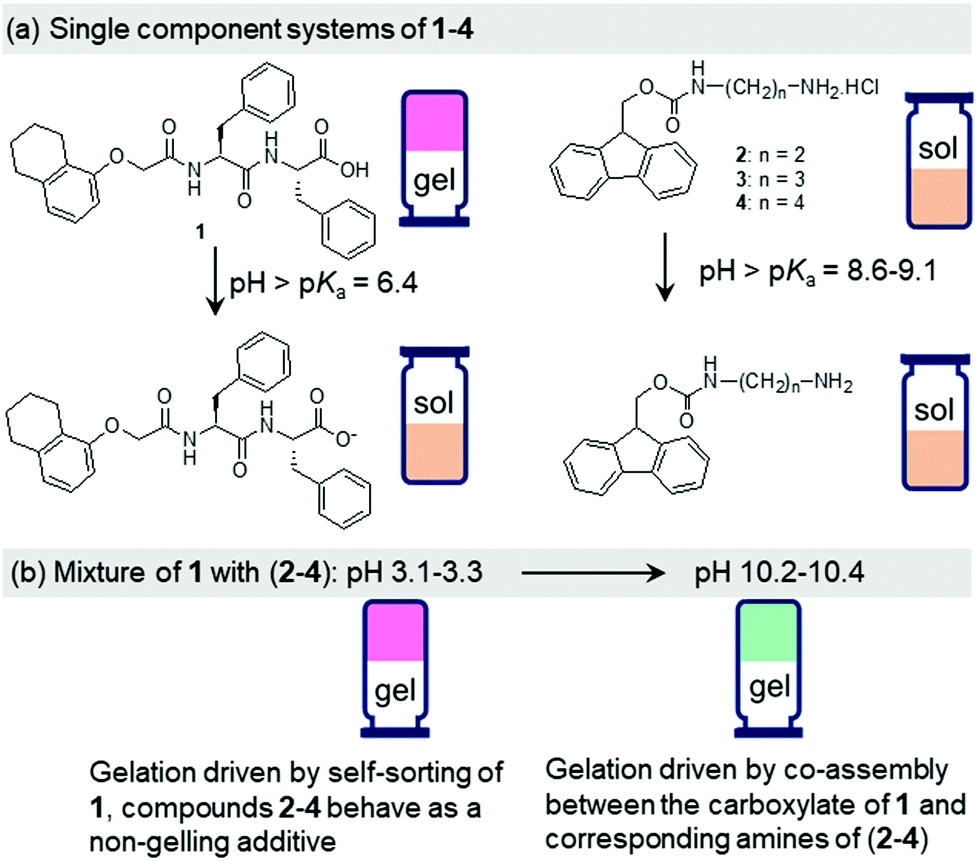
At high pH, all the single-component systems 1–4 did not have gelling ability (Fig. S9, ESI†). However, mixing of 1 with 2–4 in presence of NaOH individually resulted in a gel in all cases (pH is 10.2–10.4 in presence of 0.01 M NaOH) (Fig. S17, ESI†). Hence, at high pH, gelation was driven by the co-assembly between the components (Scheme S1, ESI†).29 Interestingly, the microstructure and the mechanical properties of the co-assembled gels are significantly influenced by the hydrophobicity of the ammonium salts. While the multicomponent gels of (1 + 2) and (1 + 3) exhibited long fibre formation, the mixed system (1 + 4) showed large spherulitic domains as the microstructure at high pH (Fig. 2). A linear increase in stiffness (G′) of the gels was realized on moving from (1 + 2) to (1 + 4) (Fig. 3a and Fig. S16, S18, ESI†). The hydrogel of (1 + 4) showed ∼25 times higher stiffness compared to the (1 + 2) gel at high pH. Despite their high stiffness however, the gels break down at relatively low strain. The gel strength (critical strain) gradually reduced from ∼13% to ∼3% on moving from the system (1 + 2) to (1 + 4).
 | ||
| Fig. 3 (a) Strain sweep experiments of the multicomponent gels prepared at pH 10.2–10.4. The black, red, and blue data represent hydrogels of (1 + 2), (1 + 3), and (1 + 4), respectively, at high pH. The closed symbols represent G′, the open symbols G′′. The dotted vertical lines represent the maximum strain bearing capacity of the corresponding hydrogel. (b) Normalized emission spectra of the multicomponent gels at pH 3.1–3.3 (dotted lines) and pH 10.2–10.4 (solid lines). The black, red, and blue data represent hydrogels of (1 + 2), (1 + 3), and (1 + 4), respectively. In all cases, initial concentrations of 1–4 are 2 mg mL−1, concentration of NaOH is 0.01 M, solvent is DMSO/H2O (20/80, v/v). | ||
The differences in the properties of the multicomponent gels at different pH can be ascribed to the change in underlying molecular packing due to the existence of different non-covalent interactions at acidic and basic pH.15 To confirm this, FTIR, UV-vis and fluorescence studies were conducted on the gels. In the FTIR spectra of 2–4, the carbamate carbonyl stretching appeared at 1687–1689 cm−1 (Fig. S19, ESI†). The self-sorted gel of 1 alone exhibited strong amide carbonyl stretching at 1648 cm−1 along with a shoulder peak at 1687 cm−1 characteristic to the formation of antiparallel β-sheet structures through intermolecular hydrogen bonding involving the amide groups (Fig. S20, ESI†).30 For all of the multicomponent gels at low pH, the amide carbonyl stretching remains almost unaffected, appearing at ∼1647 cm−1 (Fig. S21, ESI†). Interestingly, while the shoulder at 1687 cm−1 intensified and shifted to 1681 cm−1 in the multicomponent gel of (1 + 2) due to overlapping with the carbamate carbonyl of 2, it became broad in the gels of (1 + 3) and (1 + 4). Extensive broadening of the carboxylic carbonyl (appearing at 1723 cm−1 in the gel of 1 alone) as well as the amide –NHs and carboxylic –OH of 1 was also recorded in presence of 2–4. In presence of base, a substantial change in amide carbonyl stretching was realized for the multicomponent gels at high pH. In all cases, the β-sheet structures were retained at high pH with the signatures of amide and carbamate carbonyl stretching in the region 1649–1633 cm−1 (Fig. S21, ESI†).15,31 However, appearance of multiple peaks with different intensities in the range 1649–1633 cm−1 suggests co-existence of varieties of H-bonding stack sizes.31 Depending on the structural flexibility and rigidity, compounds 2–4 can attain different hydrogen bonding attributes at high pH leading to different molecular packing31 in the co-assembled gels.
The emission profiles of the multicomponent gels are identical at low pH. In all cases, emission wavelength of the Fmoc-amphiphiles merged with the emission of 1 and appeared at ∼320 nm with almost equal intensity (Fig. S12, S22 and S23, ESI†). It is notable that, unlike the compounds 2–4 (Fig. S12, ESI†), the hydrogel of 1 alone is poorly emissive and exhibits negligible changes in emission spectra on pH perturbation (Fig. S22, ESI†). For the multicomponent gels, the peak near 320 nm underwent a 10–40 nm red shift with reduced intensity on increasing the pH (Fig. S23, ESI†). Interestingly, all the high-pH gels exhibited excimer emission in the region 425–525 nm.15 The relative intensity of the excimer peaks gradually increases from the (1 + 2) to (1 + 4) systems (Fig. 3b). These observations demonstrate that aromatic stacking between the Fmoc-groups only exists at high-pH multicomponent gels.15 The degree of aromatic stacking in the co-assemblies increases with increase in the hydrophobic chain length of Fmoc-components. The peak at 300 nm in the UV-Vis spectra of low-pH multicomponent gels underwent a red shift of around 2 nm along with significant broadening at 278 nm on addition of base in all cases (Fig. S24, ESI†). Hence, comparison of spectroscopic data reveals that in the multicomponent systems, while intermolecular hydrogen bonding maintains the hydrogel networks at low pH, both aromatic stacking and H-bonding interactions control the properties of gels at high pH. Co-assembled gels with a greater extent of aromatic stacking exhibit higher stiffness in rheology (Fig. 3).
The ambidextrous phase behaviour of the multicomponent systems can be utilized to construct pH-driven self-regulating switchable materials. To execute this, we coupled our systems with the urea-urease reaction. In water, urease hydrolyses urea into ammonia. This reaction is widely used to construct various pH responsive switchable systems with a precise control over the rate of pH change.32,33 Combination of 1 with the Fmoc-amphiphiles in presence of urease and urea initially resulted in a gel at pH 3.3–3.4 with G′ being considerably greater than G′′ in all cases (Fig. 4 and Fig. S25, ESI†). The pH-time profiles showed a sigmoidal curve where at the beginning the pH change was slow. Over time, the concentration of ammonia increases, and the pH reached a plateau of 9.3 for all systems. The variation of G′, G′′ and complex viscosity with time followed a similar trend as that of pH. However, depending on the Fmoc-component, different pH-feedback was recorded during the pH change. The rate of pH change and so the change of rheological moduli and complex viscosity gradually increases from the (1 + 2) system to the (1 + 4) system. Throughout this pH change no sol formation occurs. Indeed, in all variations, the rheological moduli of the final gels were substantially higher (>3–6 times) than the initially formed materials (Fig. 4 and Fig. S26–S28, ESI†). Again, the stiffness of the high-pH gels follows the order of hydrophobicity of the Fmoc-amphiphiles (Fig. S29, ESI†). Time-variable fluorescence data showed gradual appearance of excimer peaks in the region 425–525 nm along with a 7–16 nm red shift in emission of the initial gels during the pH change (Fig. S30, ESI†). Again, the relative intensity of the excimer peaks increases in the order (1 + 2) to (1 + 4) at high pH (Fig. S31, ESI†). Furthermore, 3–4 nm red shifts in the absorption spectra of the gels intimate that there is a change in the molecular packing as the pH increases (Fig. S32, ESI†). Compared to the NaOH-triggered gels, the self-regulating approach leads to gels that are more reproducible as observed from small error bars in the rheological data (Fig. S16 and S29, ESI†). The microstructures of the high pH gels obtained by the enzymatic reaction are also different to those of the NaOH-triggered gels. Here, all the high pH gels exhibit densely packed large spherulitic domains of fibre as the underlying network (Fig. S33, ESI†).
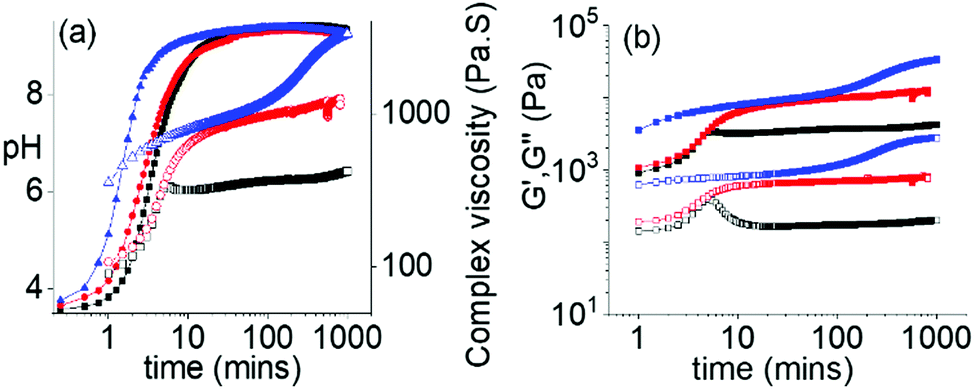 | ||
| Fig. 4 (a) Variation of pH (closed symbols) and complex viscosity (open symbols), and (b) Variation of G′ (closed symbols) and G′′ (open symbols) with time for the multicomponent systems (1 + 2) (black), (1 + 3) (red) and (1 + 4) (blue) in presence of urease–urea reaction involving initial conditions: [1–4] = 2 mg mL−1, [urease] = 0.5 mg mL−1, [urea] = 0.1 M. In all cases, solvent is DMSO/H2O (20/80, v/v). | ||
In conclusion, we demonstrated a simple method to adapt material properties of pH responsive multicomponent gels comprising of gelators with two oppositely ionizable pendant groups. We found that, when a component acts as a non-gelling additive, increasing the size of a hydrophobic structural feature on that component hardly perturbs the self-sorting of the second gelator building block at low pH. However, increasing hydrophobic chain length significantly influences the underlying molecular packing as well as microstructures and mechanical properties in co-assemblies at basic pH. Furthermore, a pH driven self-regulating switchable system is constructed by incorporating the urease–urea reaction into the multicomponent gel which allows us to achieve precise control over the final mechanical properties of the materials. These insights should be useful in devising new multicomponent hydrogels particularly with ambidextrous phase behaviour.
SP thanks the University of Glasgow for funding. BD thanks the EPSRC for funding (EP/S019472/1).
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Kollman, in New Comprehensive Biochemistry, ed. M. I. Page, Elsevier, 1984, vol. 6, pp. 55–71 Search PubMed.
- P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS.
- V. Nele, J. P. Wojciechowski, J. P. K. Armstrong and M. M. Stevens, Adv. Funct. Mater., 2020, 30, 2002759 CrossRef CAS.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- C.-W. Chu and C. A. Schalley, Org. Mater., 2021, 03, 025–040 CrossRef CAS.
- Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC.
- M. Liu, G. Ouyang, D. Niu and Y. Sang, Org. Chem. Front., 2018, 5, 2885–2900 RSC.
- P. Dastidar, R. Roy, R. Parveen and K. Sarkar, Adv. Ther., 2019, 2, 1800061 CrossRef.
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC.
- B. O. Okesola, Y. Wu, B. Derkus, S. Gani, D. Wu, D. Knani, D. K. Smith, D. J. Adams and A. Mata, Chem. Mater., 2019, 31, 7883–7897 CrossRef CAS PubMed.
- D. M. Raymond and B. L. Nilsson, Chem. Soc. Rev., 2018, 47, 3659–3720 RSC.
- L. Li, R. Sun and R. Zheng, Mater. Des., 2021, 197, 109209 CrossRef CAS.
- S. Panja, B. Dietrich, O. Shebanova, A. Smith and D. J. Adams, Angew. Chem., Int. Ed., 2021, 60, 9973–9977 ( Angew. Chem. , 2021 , 133 , 10061–10065 ) CrossRef CAS PubMed.
- D. Ghosh, A. D. Farahani, A. D. Martin, P. Thordarson and K. K. Damodaran, Chem. Mater., 2020, 32, 3517–3527 CrossRef CAS.
- C. Camilloni, D. Bonetti, A. Morrone, R. Giri, C. M. Dobson, M. Brunori, S. Gianni and M. Vendruscolo, Sci. Rep., 2016, 6, 28285 CrossRef CAS PubMed.
- A. D. Martin, J. P. Wojciechowski, A. B. Robinson, C. Heu, C. J. Garvey, J. Ratcliffe, L. J. Waddington, J. Gardiner and P. Thordarson, Sci. Rep., 2017, 7, 43947 CrossRef PubMed.
- K. McAulay, P. A. Ucha, H. Wang, A. M. Fuentes-Caparrós, L. Thomson, O. Maklad, N. Khunti, N. Cowieson, M. Wallace, H. Cui, R. J. Poole, A. Seddon and D. J. Adams, Chem. Commun., 2020, 56, 4094–4097 RSC.
- S. Ogi, V. Stepanenko, J. Thein and F. Würthner, J. Am. Chem. Soc., 2016, 138, 670–678 CrossRef CAS PubMed.
- H.-K. Yang, H. Zhao, P.-R. Yang and C.-H. Huang, Colloids Surf., A, 2017, 535, 242–250 CrossRef CAS.
- R. Chen, C. Xu, Y. Lei, H. Liu, Y. Zhu, J. Zhang and L. Xu, RSC Adv., 2021, 11, 12641–12648 RSC.
- Q. Zhang, M. Wu, X. Hu, W. Lu, M. Wang, T. Li and Y. Zhao, Macromol. Chem. Phys., 2020, 221, 1900320 CrossRef CAS.
- C. Berdugo, J. F. Miravet and B. Escuder, Chem. Commun., 2013, 49, 10608–10610 RSC.
- Y. Gao, F. Zhao, Q. Wang, Y. Zhang and B. Xu, Chem. Soc. Rev., 2010, 39, 3425–3433 RSC.
- S. Panja, A. M. Fuentes-Caparrós, E. R. Cross, L. Cavalcanti and D. J. Adams, Chem. Mater., 2020, 32, 5264–5271 CrossRef CAS PubMed.
- S. Debnath, S. Roy, Y. M. Abul-Haija, P. W. J. M. Frederix, S. M. Ramalhete, A. R. Hirst, N. Javid, N. T. Hunt, S. M. Kelly, J. Angulo, Y. Z. Khimyak and R. V. Ulijn, Chem. – Eur. J., 2019, 25, 7881–7887 CrossRef CAS PubMed.
- S. Roy, N. Javid, J. Sefcik, P. J. Halling and R. V. Ulijn, Langmuir, 2012, 28, 16664–16670 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- J. D. Tang, C. Mura and K. J. Lampe, J. Am. Chem. Soc., 2019, 141, 4886–4899 CrossRef CAS PubMed.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, T. Tuttle and R. V. Ulijn, Chem. Commun., 2013, 49, 10587–10589 RSC.
- E. Jee, T. Bánsági Jr., A. F. Taylor and J. A. Pojman, Angew. Chem., Int. Ed., 2016, 55, 2127–2131 CrossRef CAS PubMed.
- S. Panja and D. J. Adams, Chem. – Eur. J., 2021, 27, 8928–8939 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc02786g |
| This journal is © The Royal Society of Chemistry 2021 |