
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalyzed sp–sp3 cross-coupling of benzyl bromides using lithium acetylides†
Anirban
Mondal
 ,
Paco
Visser
,
Anna M.
Doze
,
Jeffrey
Buter
* and
Ben L.
Feringa
*
,
Paco
Visser
,
Anna M.
Doze
,
Jeffrey
Buter
* and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, Groningen 9747 AG, The Netherlands. E-mail: j.buter@rug.nl; b.l.feringa@rug.nl
First published on 1st July 2021
Abstract
Organolithium-based cross-coupling reactions have emerged as an indispensable method to construct C–C bonds. These transformations have proven particularly useful for the direct and fast coupling of various organolithium reagents (sp, sp2, and sp3) with aromatic (pseudo) halides (sp2). Here we present an efficient method for the cross-coupling of benzyl bromides (sp3) with lithium acetylides (sp). The reaction proceeds within 10 min at room temperature and can be performed in the presence of organolithium-sensitive functional groups such as esters, nitriles, amides and boronic esters. The potential application of the methodology is demonstrated in the preparation of key intermediates used in pharmaceuticals, chemical biology and natural products.
Benzyl alkynes are versatile synthetic intermediates,1 which are also found in several natural products,2 pharmaceuticals, polymeric materials3 and applied in click chemistry.4 Although, at first sight, alkynylation at the benzylic position appears trivial, this transformation has proven to be surprisingly challenging as exemplified in previous reports showing the necessity of (i) transition metal-catalyzed cross-coupling of metal-alkynyls;5 (ii) Sonogashira-type coupling using terminal alkynes;6 and (iii) direct substitution of benzyl halides with an alkynyl lithium in the presence of the deaggregation agent N,N′-dimethylpropyleneurea (DMPU).7 Despite the intrinsic reactivity of both the nucleophile and electrophile in the latter example, this ‘textbook’ SN2 reaction required DMPU to proceed.8 This method can therefore not be applied in the presence of organolithium-sensitive functional groups due to the enhanced reactivity of organolithium reagents in the presence of e.g. DMPU, HMPA, TMEDA.9 Following pioneering work by Murahashi, on the early development of organolithium-based cross-coupling reactions,9,10 our group introduced a general methodology for the direct palladium-catalyzed cross-coupling of organolithium reagents with aryl- and alkenyl bromides to construct C–C bonds,11 thereby avoiding transmetalation to e.g. organozinc, –boron, –silicon, or –tin reagents. Having successfully tamed the extreme reactivity of organolithium compounds12 in batch reactions, these palladium-catalyzed C–C bond formation reactions proceed fast (5–60 min) with high conversion and selectivity,13 and can be performed at room temperature,14 with a nickel catalyst,15 without solvent,16 under air,16 on ‘water’,17 at low temperature18 and have found application in stereoselective natural product synthesis.19 Recently, we reported a Sonogashira coupling employing lithium acetylides.20 These reactions proceed at room temperature within 45 min, and display a remarkable functional group tolerance, including organolithium-sensitive functional groups such as esters, nitriles and boronic esters. Having demonstrated the cross-coupling of lithium acetylides with aryl bromides, we envisioned the development of fast and selective methodology for the functionalization of benzyl bromides (Scheme 1). Herein, we describe the cross-coupling of a variety of benzyl bromides with lithium acetylides, proceeding at room temperature in only ten minutes. Notably, the protocol shows high selectivity and an impressive tolerance towards functional groups such as esters, amides, nitriles, and a boronic ester. Furthermore, the versatility of the benzylic alkynes was displayed in the construction of key intermediates applied in the synthesis of pharmaceuticals and natural products.
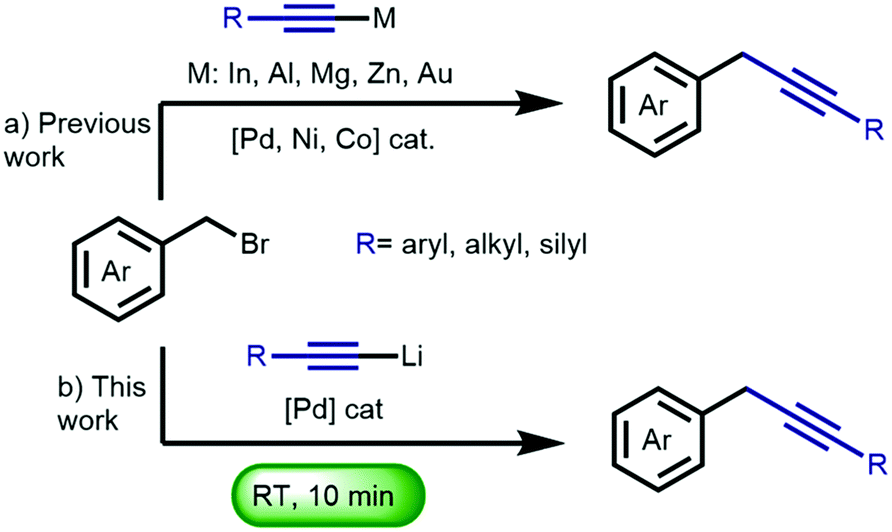 | ||
| Scheme 1 (a) Previous approaches towards the transition metal-catalyzed cross-coupling of organometallic acetylenes with benzyl bromides. (b) This work. | ||
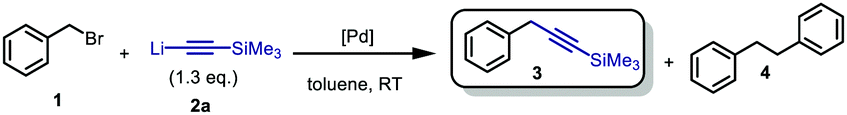
Our investigations started by reacting benzyl bromide, as well as its p-OMe and p-CF3 derivatives, with trimethylsilyl- and tri(isopropyl)-silyl lithium acetylide, using different solvents, which in the absence of a transition-metal catalysts were affording only mediocre conversion to 3 (16%, THF after 2 h, see ESI† Table S1).
Moreover, performing the reaction in N-methyl-2-pyrrolidone (NMP), DMPU, or toluene also did not result in a productive reaction. These initial observations clearly demonstrated the necessity for the development of a catalytic method. Next the screening of a series of different Pd-catalysts and varying the addition time of the lithium acetylide 2a (Table 1) was pursued. Satisfyingly, the use of 5 mol% of oxygen-activated Pd[P(tBu3)]2,14 a catalyst we previously disclosed to be effective for the Sonogashira-type cross-coupling of lithium acetylides with aryl bromides,20 resulted in 98% conversion into the desired product 3 (entry 1). Lowering the addition time from 30 to 15 min retained quantitative conversion, albeit with slightly lower selectivity (95%, entry 2). Moderate conversion of 44%, paired with a decreased selectivity, was observed when we lowered the addition time from 15 to 5 min (entries 2 vs. 3). Next, we investigated the use of [Pd(μ-I)PtBu3]2, previously reported as an effective catalyst in Kumada-type cross-couplings by Schoenebeck and co-workers.21 Addition of the lithium acetylide over either 10 or 5 min in the presence of 5 mol% catalyst resulted in near-quantitative conversion with excellent selectivity (entries 4 and 5). Decreasing the catalyst loading to 2.5 mol% with an addition time of 5 min, however resulted in moderate conversion with decreased selectivity (entry 6). After optimizing the addition time, as well as the reaction conditions and organolithium-concentration, we found that optimal results were obtained using 3 mol% of the Pd–I dimer complex with toluene as solvent, and an addition time of 10 min of the lithium acetylide (see ESI† Table S1). With a growing incentive to develop more sustainable chemical reactions, in which the amount of waste generated is minimized,22 we were also keen to explore whether this reaction could be performed without the use of additional solvents. Diminished solvent use could result in a transformation with a decreased E-factor.23 Addition of 2a to a mixture of 1 and Pd[P(tBu)3]2 over 5 min under open-flask conditions provided the desired product 3 with 74% conversion (entry 8). Increasing the catalyst loading from 5 to 7.5% greatly improved both conversion and selectivity (entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Mol% | Time (min) | Conversion of 1 (%) |
3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4b (%) :4b (%) |
| Reaction conditions.a RLi prepared from trimethylsilyl acetylene (0.65 mmol) and nBuLi (1.6 M in hexanes; 1 equiv.) in THF (0.5 M) was added dropwise to a solution of [Pd] and 1 (0.5 mmol) in toluene (4 mL).b Conversion determined by GC–MS analysis.c RLi was prepared from trimethylsilyl acetylene (0.65 mmol) and nBuLi (1.6 M in hexanes; 1 equiv.) in THF (0.6 M), and diluted with toluene (final concentration: 0.35 M).d The reaction was performed under air in the absence of additional solvent. | |||||
| 1 | Pd(PtBu3)2/O2 | 5 | 30 | >99 | 98:2 |
| 2 | 15 | >99 | 95:5 |
||
| 3 | 5 | 44 | 82:18 |
||
| 4 | [Pd(μ-I)PtBu3]2 | 5 | 10 | 98 | 97:3 |
| 5 | 5 | 99 | 96:4 |
||
| 6 | [Pd(μ-I)PtBu3]2 | 2.5 | 5 | 59 | 88:12 |
| 7 | [Pd(μ-I)P t Bu 3 ]2c | 3 | 10 | >99 |
96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :4 :4 |
| 8 | Pd(PtBu3)2/under aird | 5 | 5 | 87 | 86:14 |
| 9 | Pd(PtBu3)2/under aird | 7.5 | 5 | >99 | 94:6 |
Although promising in terms of low E-factor, further screening showed that the general applicability was more troublesome, presumably due to the heterogenic nature of the reaction. With the optimized conditions obtained, using [Pd(μ-I)PtBu3]2 (3 mol%) as the catalyst (see ESI† Fig. S1 for the proposed mechanism), we continued to investigate the general nature of the transformation by reacting benzyl bromides bearing different arene substituents (Scheme 2). During our initial studies, we found that chromatographic separation of the desired products from minor amounts of homo-coupling side-products proved to be challenging. However, we discovered that simply switching from lithium (trimethylsilyl)acetylide 2a to lithium(triisopropylsilyl) acetylide 2b circumvented this issue. Hence, we evaluated the generality of the reaction mainly using 2b. Benzyl bromides with various arene substituents reacted smoothly towards the corresponding coupling products. Unsubstituted benzyl bromide was converted with both 2a and 2b to form 3 and 5 in excellent yield. Performing the synthesis of 5 on gram-scale afforded the desired product in similar yield (82%). The reaction of 2-(bromomethyl)-naphthalene also proceeded, providing 6 in 62% yield. Substrates bearing p-alkyl substituents readily underwent the cross-coupling reaction, to give 7 and 8 in 91% and 70% yield, respectively. The presence of electron-donating para-substituents was also tolerated i.e. p-thioanisole 9 (81%). Substituents in the meta-position also posed no problem since m-methoxy- and m-methyl-substituted benzyl bromides were efficiently converted to the corresponding products 10 (84%) and 11 (86%), respectively. Sterically demanding o-substituted methyl- and phenyl benzyl bromides remained challenging, resulting in non-productive reactions with lithium(triisopropylsilyl) acetylide 2b. Reducing the steric bulk of the acetylide by using lithium (trimethylsilyl) acetylide (2a) proved crucial, and smoothly provided 12 and 13 (see ESI† Fig. S1 for detailed explanation). Next, we moved our attention towards electron-poor benzyl bromides. The reaction of both m and p-CF3 benzyl bromide provided the desired product 14 and 15 in 61% and 75% yield, respectively. A significant difference in reactivity, however, was observed in the reactions of p- and m-nitrobenzyl bromide. The attempted cross-coupling with p-nitrobenzyl bromide proceeded with full conversion towards the homo-coupled 1,2-biarylethane. Due to the high acidity of the benzylic position (pKap-nitrotoluene = 20.4 vs. pKa acetylene = 25),24 lithium–halogen exchange results in the formation of a more stable p-nitrobenzyl anion, subsequently forming the 1,2-biarylethane side-product.25 As expected, the homo-coupling product was not observed when we moved the nitro-substituent out of conjugation with respect to the benzylic position, which provided us with the desired product 17 in 45% yield. Although organolithium reagents can be of great use, their inherent reactivity generally might limit their effectiveness as cross-coupling reagents in the presence of functional groups. In the case of lithium acetylides, due to their lower pKa, and therefore diminished reactivity compared to alkyl- and alkenyllithium reagents, we anticipated that this transformation should be feasible in the presence of organolithium-sensitive functionalities. A nitrile-bearing substrate, which is prone to mono- or double addition reactions with organolithium reagents to form various imines and carbinamines,26 could be efficiently transformed into coupled product 18 (75%). The presence of amides and esters are well tolerated and furnish 19–21 in high yields, despite their usual tendency to engage in undesired 1,2-addition reactions with organolithium reagents to form ketones and alcohols. Furthermore, the presence of a boronic ester, a versatile functionality for Suzuki–Miyaura cross-coupling reactions, was well-tolerated, despite the fact that they easily react with organolithium compounds to form boronates.27 Under our reaction conditions, bifunctional product 22 was readily synthesized (54%). A particularly challenging substrate is phenylacetonitrile-based benzyl bromide, containing, in addition to the reactive nitrile functionality, also an acidic position (pKa of phenylacetonitrile ∼22).24 Despite bearing these two reactive sites, our procedure afforded 23 in 47% yield. The use of the azobenzene moiety in photopharmacology28 is of great interest due to its easy access and photo-switching behavior. The introduction of the benzylic alkyne functionality allows for facile attachment of the pharmacophore by means of Cu-catalyzed click-chemistry.4,29 Using our method, we successfully attached the ‘click handle’ to the azobenzene core in 67% yield (24). We also examined the reactions of benzyl bromides bearing both C(sp2)- and C(sp3)-bromides, as we previously observed that [Pd(μ-I)PtBu3]2 is also an effective catalyst for the cross-coupling of lithium acetylides with aryl bromides.20 We were pleased to obtain the double coupling products 25–27 in 48–52% yield. These products are highly versatile synthons due to their application in various cycloaddition and cyclisation reactions.30 Moreover, we were also able to synthesize di-(28) and tri-(29) substituted benzyl acetylenes in a one-pot procedure providing excellent yields of multiple coupled products. These compounds can fulfill roles as bridging/spacer units and dendrimer building blocks (core and branching point), respectively.31 Finally, we also performed the cross-coupling reactions by reacting two other substituted lithium acetylides with m-ester substituted benzyl bromide. As expected, the lithium acetylenes generated from both phenylacetylene and 9-ethynylphenanthrene reacted smoothly and afforded 30–31 in 46% and 52% yield. In order to demonstrate the potential synthetic application of the methodology, we envisioned benzylic alkynes as versatile precursors for a wide array of products.3 There are numerous applications of the terminal alkynes3 which are directly accessible via our cross-coupling product using a TBAF-mediated deprotection.32 It should be noted that benzylic alkynes can undergo alkyne-allene isomerization under basic deprotection conditions. This can however be circumvented by carrying out the reaction with AgNO3/KI.33 Importantly however, allenes can also be very useful functional groups in view of their reactivity towards cycloaddition and cyclisation reaction.34 We compared the use our new acetylene coupling reaction to previously reported syntheses of important benzylic alkynes (Scheme 3). Fluorobenzyl alkyne 32 was used for the synthesis of CRTH2 receptor antagonists 33.35 The reported approach required stoichiometric amounts of CuI, and a reaction time of 24 h at 75 °C.36 Our methodology provided 32 in merely 10 min at room temperature, in a comparable yield (75% vs. 78%). Furthermore, benzyl alkyne 34 is of particular interesting since it is a common precursor used in the synthesis of lignans. These are plant-based natural products well-known for a wide variety of biological activities such as antitumor, antimitotic, and antiviral properties.37 Interestingly, coupling product 34 has been used as a building block for the synthesis of metabolically stable steganacin analogue 35.38 This natural product derived compound exhibited cytotoxic activity against neuroblastoma cells. The reported procedure required a reaction time of 16 h at 66 °C, providing the desired product in 90% yield. Our method gave rise to the product in slightly reduced yield (82%), albeit using much milder (RT) and significantly more time-efficient (30 min) conditions.
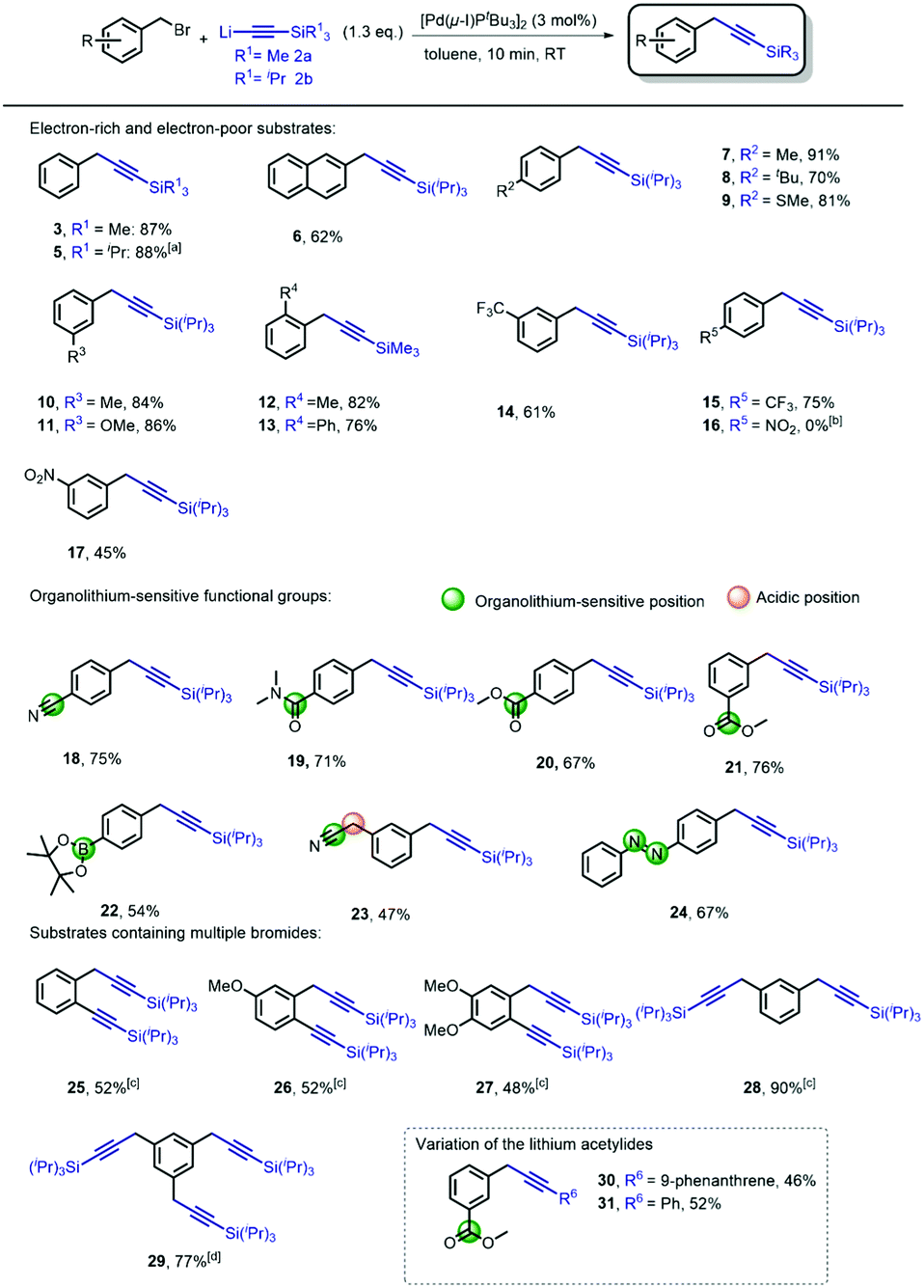 | ||
| Scheme 2 Scope of the benzyl alkyne synthesis. Reaction conditions. Freshly prepared 2b (1.3 equiv.; 0.35 M) was added to a solution, of [Pd(μ-I)PtBu3]2 (3 mol%) and corresponding benzyl bromide (0.5 mmol) in toluene (2 mL), over 10 min. [a] The reaction was also performed on 1 gram scale with 82% yield [b] Quantitative formation to corresponding 1,2-biarylethane [c] Freshly prepared 2b (2.6 equiv.) was added to a solution of [Pd(μ-I)PtBu3]2 (6 mol%) and 1 (0.5 mmol) in toluene (4 mL) over 20 min. [d] 3.9 equiv. of 2b was used (also see ESI† for more information). | ||
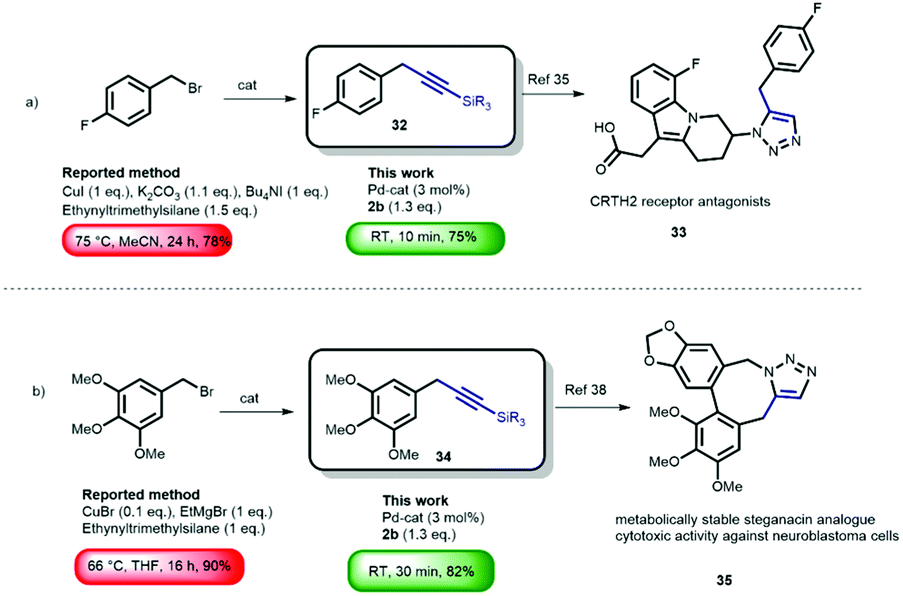 | ||
| Scheme 3 Potential applications of the cross-coupling methodology. A comparison of the developed protocol with reported syntheses of different drugs. | ||
In summary, we have developed a mild, fast and highly selective approach for the catalytic cross-coupling of benzyl bromides with lithium acetylides. The reaction proceeds in ten minutes at room temperature, offering an efficient pathway to a range of protected benzylic acetylenes in generally high yields under mild conditions. Due to the remarkable functional group tolerance of the transformation, the alkynylation of benzyl bromides with lithium acetylides might likely find applications in the late-stage functionalization of (photo-)pharmacophores, natural products, materials and pharmaceuticals.
This work was financially supported by ERC (advanced grant No. 694345) and the Dutch Ministry of Education, Culture and Science (Gravitation program No. 024.001.035) to B. L. F and N. W. O. (Grant Number: 718,015,004) to A. M. D. Kolarski is acknowledged for providing (E)-1-(4-(bromomethyl)-phenyl)-2-phenyldiazene. R. Sneep is acknowledged for performing the high-resolution mass spectrometry.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- B. Liégault, J.-L. Renaud and C. Bruneau, Chem. Soc. Rev., 2008, 37, 290–299 RSC.
- X. Zhu, J. Liu and W. Zhang, Nat. Chem. Biol., 2015, 11, 115–120 CrossRef CAS PubMed.
- J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS PubMed.
- (a) O. Di Pietro, N. Alencar, G. Esteban, E. Viayna, N. Szałaj, J. Vázquez, J. Juárez-Jiménez, I. Sola, B. Pérez, M. Solé, M. Unzeta, D. Muñoz-Torrero and F. J. Luque, Bioorg. Med. Chem., 2016, 24, 4835–4854 CrossRef; (b) F. Li, Y. Park, J.-M. Hah and J.-S. Ryu, Bioorg. Med. Chem. Lett., 2013, 23, 1083–1086 CrossRef CAS PubMed.
- (a) M. Qian and E.-i. Negishi, Tetrahedron Lett., 2005, 46, 2927–2930 CrossRef CAS; (b) I. Pérez, J. P. Sestelo and L. A. Sarandeses, J. Am. Chem. Soc., 2001, 123, 4155–4160 CrossRef PubMed; (c) M. Peña-López, M. Ayán-Varela, L. A. Sarandeses and J. Pérez Sestelo, Chem. – Eur. J., 2010, 16, 9905–9909 CrossRef PubMed; (d) D. B. Biradar and H.-M. Gau, Chem. Commun., 2011, 47, 10467–10469 RSC; (e) A. Kuno, N. Saino, T. Kamachi and S. Okamoto, Tetrahedron Lett., 2006, 47, 2591–2594 CrossRef CAS.
- (a) H. Zhang, N. Sun, B. Hu, Z. Shen, X. Hu and L. Jin, Org. Chem. Front., 2019, 6, 1983–1988 RSC; (b) X.-Y. Dong, Y.-F. Zhang, C.-L. Ma, Q.-S. Gu, F.-L. Wang, Z.-L. Li, S.-P. Jiang and X.-Y. Liu, Nat. Chem., 2019, 11, 1158–1166 CrossRef CAS PubMed.
- T. Takahashi, M. Kitamura, B. Shen and K. Nakajima, J. Am. Chem. Soc., 2000, 122, 12876–12877 CrossRef CAS.
- C. Ruiz, Á. Raya-Barón, M. A. Ortuño and I. Fernández, Dalton Trans., 2020, 49, 7932–7937 RSC.
- U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194–204 CrossRef CAS.
- For key contribution in the development of organolithium-based cross-coupling please see; (a) S. Murahashi, M. Yamamura, K. Yanagisawa, N. Mita and K. Kondo, J. Org. Chem., 1979, 44, 2408–2417 CrossRef CAS; (b) A. Nagaki, A. Kenmoku, Y. Moriwaki, A. Hayashi and J. Yoshida, Angew. Chem., Int. Ed., 2010, 49, 7543–7547 CrossRef CAS PubMed; (c) E.-i. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed; (d) A. B. Smith, W.-S. Kim and R. Tong, Org. Lett., 2010, 12, 588–591 CrossRef CAS PubMed.
- M. Giannerini, M. Fananas-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667–672 CrossRef CAS PubMed.
- (a) R. Luisi and V. Capriati, Lithium Compounds in Organic Synthesis, Wiley-VCH, Germany, 2014 CrossRef; (b) Z. Rappoport and I. Marek, The chemistry of organolithium compounds, John Wiley & Sons, 2004 CrossRef.
- J. Mateos-Gil, A. Mondal, M. Castiñeira Reis and B. L. Feringa, Angew. Chem., Int. Ed., 2020, 59, 7823–7829 CrossRef CAS PubMed.
- D. Heijnen, F. Tosi, C. Vila, M. C. A. Stuart, P. H. Elsinga, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 3354–3359 CrossRef CAS PubMed.
- D. Heijnen, J.-B. Gualtierotti, V. Hornillos and B. L. Feringa, Chem. – Eur. J., 2016, 22, 3991–3995 CrossRef CAS PubMed.
- E. B. Pinxterhuis, M. Giannerini, V. Hornillos and B. L. Feringa, Nat. Commun., 2016, 7, 11698 CrossRef PubMed.
- G. Dilauro, A. Francesca Quivelli, P. Vitale, V. Capriati and F. M. Perna, Angew. Chem., Int. Ed., 2019, 58, 1799–1802 CrossRef CAS PubMed.
- N. Sinha, D. Heijnen, B. L. Feringa and M. G. Organ, Chem. – Eur. J., 2019, 25, 9180–9184 CrossRef CAS PubMed.
- J. Buter, D. Heijnen, C. Vila, V. Hornillos, E. Otten, M. Giannerini, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 3620–3624 CrossRef CAS PubMed.
- H. Helbert, P. Visser, J. G. H. Hermens, J. Buter and B. L. Feringa, Nat. Catal., 2020, 3, 664–671 CrossRef CAS.
- F. Proutiere, E. Lyngvi, M. Aufiero, I. A. Sanhueza and F. Schoenebeck, Organometallics, 2014, 33, 6879–6884 CrossRef CAS.
- (a) R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC; (b) M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807–810 CrossRef CAS PubMed; (c) J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- K. Izutsu, Acid-base Dissociation Constants in Dipolar Aprotic Solvents, Blackwell Scientific Publications, 1990 Search PubMed.
- E. B. Pinxterhuis, P. Visser, I. Esser, J.-B. Gualtierotti and B. L. Feringa, Angew. Chem., Int. Ed., 2018, 57, 9452–9455 CrossRef CAS PubMed.
- M. S. M. Pearson-Long, F. Boeda and P. Bertus, Adv. Synth. Catal., 2017, 359, 179–201 CrossRef CAS.
- C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- H. Chen, R. Zhang, H. Gao, H. Cheng, H. Fang and X. Cheng, Dyes Pigm., 2018, 149, 512–520 CrossRef CAS.
- (a) R. P. Kaiser, F. Hessler, J. Mosinger, I. Císařová and M. Kotora, Chem. – Eur. J., 2015, 21, 13577–13582 CrossRef CAS PubMed; (b) M. Mandal, S. Sakthivel and R. Balamurugan, J. Org. Chem., 2021, 86, 333–351 CrossRef CAS PubMed.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed.
- J. A. Fernández-Salas, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2016, 138, 790–793 CrossRef PubMed.
- (a) H. M. Schmidt and J. F. Arens, Recl. Trav. Chim. Pays-Bas, 1967, 86, 1138–1142 CrossRef CAS; (b) L. K. Geisler, S. Nguyen and C. J. Forsyth, Org. Lett., 2004, 6, 4159–4162 CrossRef CAS PubMed.
- (a) F. López and J. L. Mascareñas, Chem. Soc. Rev., 2014, 43, 2904–2915 RSC; (b) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2014, 43, 3106–3135 RSC.
- C. Berthelette, M. Boyd, J. Colucci, K. Villeneuve and J. L. Methot, US. Patent., No. US9023864B2, 2015 Search PubMed.
- T. Li and L. Zhang, J. Am. Chem. Soc., 2018, 140, 17439–17443 CrossRef CAS PubMed.
- T. Umezawa, Phytochem. Rev., 2003, 2, 371–390 CrossRef CAS.
- D. Imperio, T. Pirali, U. Galli, F. Pagliai, L. Cafici, P. L. Canonico, G. Sorba, A. A. Genazzani and G. C. Tron, Bioorg. Med. Chem., 2007, 15, 6748–6757 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc02762j |
| This journal is © The Royal Society of Chemistry 2021 |