
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the stability of the NHC–metal bond using thiones as probes†
Nathalie
Ségaud
 ,
Chloë
Johnson
,
Albert
Farre
and
Martin
Albrecht
*
,
Chloë
Johnson
,
Albert
Farre
and
Martin
Albrecht
*
Department of Chemistry & Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland. E-mail: martin.albrecht@unibe.ch
First published on 10th September 2021
Abstract
The metal–carbon bond in N-heterocyclic carbene (NHC) metal complexes, which are ubiquitous in modern homogeneous catalysis, is often conjectured to be robust. Here, carbene dissociation was evaluated from a series of complexes with metals of relevance in catalysis containing either an Arduengo-type 2-imidazolylidene or a mesoionic 1,2,3-triazolylidene ligand through thione formation, revealing remarkable kinetic lability of the NHC–metal bond for, e.g. IrIII, RhIII, and NiII complexes.
N-Heterocyclic carbenes (NHC)1 have become an ubiquitous and versatile class of ligands in organometallic chemistry,2,3 largely due to their impact as powerful spectator ligands in the development of NHC metal catalysts.4–6 Especially when coordinated to late d-block metals, NHC ligands have boosted the activity of homogeneous catalysts in a broad range of applications.7–9 Generally and often by default, the extraordinary activity imparted by NHCs has been attributed to the strong σ donor properties and to the high robustness of the NHC–metal bond, which is assumed because of the relatively high degree of covalent character of the M–CNHC bond. This bonding model has been supported by several theoretical investigations,10–12 though relevant work has advised caution when conjecturing robust NHC coordination to the metal centre throughout a catalytic process.
In an early account, Crudden critically reviewed the stability of the metal–ligand bond of NHC complexes comprised of the most popular NHC ligands,13i.e. Arduengo-type imidazol-2-ylidenes.14–17 An array of decomposition pathways has been delineated including NHC ligand dissociation and substitution, migratory insertion and reductive elimination processes, which are dependent on the applied reaction conditions. More recently, Hahn and coworkers have used NHCs for supramolecular assemblies, which implies a thermodynamic control and hence reversible M–NHC bond formation.18,19 Some of these reactivity patterns have also been observed in complexes bearing 1,2,3-triazol-4-ylidenes,20 a subclass of mesoionic carbenes that emerged during the last decade.21 Degradation of some complexes was observed that involved carbene dissociation. Moreover, recent work pointed to an increased relevance of NHC dissociation for catalyst activation with a variety of metals.22,23 Here, we report on a systematic study to determine the stability of the NHC–metal bond. Specifically, we investigated a series of NHC metal complexes on their intrinsic stability, with a particular focus on potential carbene dissociation, which was monitored by trapping the released free carbene with elemental sulfur to form the corresponding thione (ESI†) as a readily detectable indirect probe.24–26 To this end, a range of NHC complexes were prepared with group 8–11 metals, as these metals are by far the most frequently used in catalytic applications, including Au(I), Pd(II), Rh and Ir both in 1+ and 3+ oxidation states, Ru(II), Ni(II), and Ag(I), as well as less explored Pt(II) and Os(II) complexes (Fig. 1). The NHC ligands used were N,N′-dibutyl-imidazol-2-ylidene (imi) as a representative of the Arduengo-type NHCs, and 1,4-dibutyl-substituted 1,2,3-triazol-4-ylidene (trz) as a mesoionic carbene (Fig. 1). The identical set of wingtip groups minimises any steric bias between these two ligand systems and allows for electronic ligand effects to be compared directly. Since the majority of catalytic systems feature more stabilizing Mes or Dipp wingtip groups, also selected IMes complexes were investigated (IMes = 1,3-dimesityl-2-imidazolylidene).
 | ||
Fig. 1 Complexes used in this study, bearing NHC ligands imi (1,3-(di-n-butyl)-imidazol-2-ylidene) or trz (1,4-(di-n-butyl)-3-methyl-1,2,3-triazolylidene). a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Dynamic mixture of [Ag(NHC)2Cl] and [Ag(NHC)Cl]. Dynamic mixture of [Ag(NHC)2Cl] and [Ag(NHC)Cl]. | ||
Silver NHC complexes were used to validate the sulfur quenching methodology, as these complexes are known to undergo rapid carbene transfer for transmetalation and also internal carbene transfer through establishing an equilibrium between neutral (NHC)AgX and ionic [(NHC)2Ag](AgX2) species in solution (Scheme 1).27,28 Indeed, in the presence of 0.6 eq. S8, complex Ag-imi transformed quantitatively into the corresponding thione imi = S within 2 h at room temperature (1,2-dichlorobenzene solution, Scheme 1 and Fig. S19, ESI†) as indicated by a distinct shift of the N–CH2 resonances from 4.09 to 4.02 ppm in the 1H NMR spectrum (CD2Cl2). Similarly, the analogous thione trz = S formed quantitatively from Ag-trz (δNCH2 = 4.45 vs. 4.41; Fig. S20, ESI†). The reaction is faster with Ag-imi than with the trz analogue (89% vs. 55% conversion after 30 min; Table S1 and Fig. S1, ESI†), though after 2 h, both complexes are quantitatively transformed into the corresponding thione.
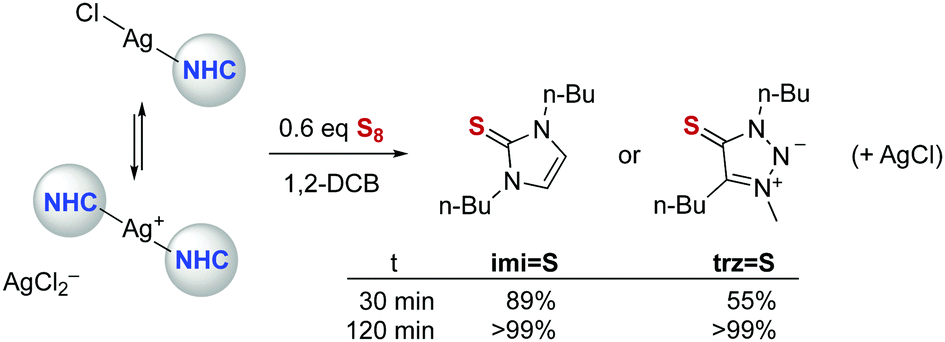 | ||
| Scheme 1 Formation of thiones imi = S and trz = S from the corresponding silver(I) NHC complexes, revealing slightly higher stability of the Ag-trz complex. | ||
Based on this validation with the silver(I) complexes, we investigated the stability of a variety of M-imi complexes. No thione formation was observed with any of the investigated group 8–11 metal complexes at RT within 24 h, apart from Ag-imi as discussed above. However, slow degradation was observed with some complexes, e.g., gradual dissociation of the spectator cymene from Ru-imi and especially COD from the only poorly stable Ir(I)-imi, as noted by NMR spectroscopy (Fig. S21, S22 and Table S2, ESI†). Decomposition was significant for Rh(I)-imi and higher for the nickel(II) and osmium(II) complexes (Fig. S23–S25, ESI†). At room temperature, the M–NHC bond is thus stable towards dissociation except for NHC–Ag complexes.
When solutions of the complexes were heated to 120 °C, however, more distinct stability profiles became apparent, including (i) thione and imidazolium salt formation because of NHC dissociation, (ii) complex decomposition without thione formation, and (iii) dissociation reactions followed by thione formation (Scheme 2). Only the gold(I) complex Au-imi did not change at all and was completely stable for 24 h (Fig. 2, Table S3 and Fig. S33, ESI†). Imi complexes with PdII, PtII, and IrI underwent degradation, yet without any formation of thione or imidazolium salt, suggesting transformations involving the ancillary ligands of the complexes without M–Cimi bond cleavage. The degradation is slow for Pd-imi and Pt-imi (7% and 31%, respectively, within 24 h), and faster for Ir(I)-imi (100% in 6 h; Fig. S34–S37, ESI†). Imi complexes of OsII, RuII, and RhI revealed some thione by NMR spectroscopy, though only after apparent modification of the initial complex, indicating labilisation of the M–Cimi bond after modification of the ancillary ligands. For example, heating Os-imi revealed loss of cymene after 30 min (13%) and 55% degradation after 24 h, but only 8% thione was formed (Fig. S39, ESI†). Similar reactivity was observed for Ru-imi (24% thione formed), while Rh(I)-imi lost COD much faster (78% decomposition after 30 min with 15% thione after 24 h, Fig. S37 and S38, ESI†). Finally, complexes Rh(III)-imi, and Ni-imi produced considerable quantities of imidazolium salt and over time also thione (48% and full degradation, respectively, after 30 min), commensurate with the degradation of the original complex and pointing to carbene dissociation under these conditions (Fig. S41 and S42, ESI†). The same process was observed for Ir(III)-imi, yet considerably slower (7% dissociation after 30 min; Fig. S40, ESI†). Indeed, a separate experiment demonstrated that the imidazolium salt [imi-H]I is gradually transformed to the thione imi = S in the presence of S8 (30% conversion in 24 h; Fig. S43, ESI†). Conversely, degradation in the absence of S8 is considerably slower, as demonstrated with Rh(III)-imi (Fig. S57, ESI†), which may point to a fast rebound of the dissociated carbene, though a sulfur-induced decomposition cannot be ruled out either.
 | ||
| Scheme 2 NHC dissociation pathways in M-imi complexes leading to either thione imi = S or imidazolium salt [imi-H]+ and complex degradation via ancillary ligand dissociation. | ||
 | ||
| Fig. 2 Amount of M-imi thione, imidazolium salt, and degradation products after 24 h reaction at 120 °C. | ||
The different behaviour of Ni-imi, Rh(III)-imi and also Ir(III)-imi compared to Ag-imi through formation of predominantly imi-H+ from the former complexes as opposed to imi = S from the silver carbene suggests that the NHC dissociation from Ag-imi is induced by sulfur binding to the metal, while in the former, NHC dissociation is assumed to be more spontaneous, with the free carbene quenched by H+ and to some extent by sulfur.
While the lability of Ag-imi and Ni-imi is not surprising since both systems have been used in transmetallation, i.e. carbene transfer reactions, the lability of Rh(III)-imi and Ir(III)-imi is remarkable. Presumably, the substantial carbene dissociation from the metal centre, as indicated by the formation of imi = S and [imi-H]+, may, in parts, originate from the mismatch of the soft carbene ligand with the relatively hard d6 metal configuration. Consistent with this consideration, also the Ru(II) complex reveals considerable carbene dissociation.
Modification of the N-substituent of the carbene from butyl to the catalytically more frequently used mesityl group has been investigated with a selection of metals which were relatively poorly stable as imi variants, namely Ag-IMes, Ni-IMes, Rh(I)-IMes, and Ir(I)-IMes. The stability profile of these complexes showed surprisingly little differences to their imi analogues (Fig. S53–S56 and Table S4, ESI†). Ir(I)-IMes is slightly more stable than Ir(I)-imi, whereas this trend is opposite for Ni(II).
Expansion of these stability studies to mesoionic 1,2,3-triazolylidene complexes20–22 reveals similar stability properties at room temperature, and thione formation was observed only with complex Ag-trz (Table S5, ESI†). The Rh(III), Ir(III), and Pd-trz complexes are completely stable over 24 h. Other complexes degraded slowly over time without formation of thione, involving either dissociation of ancillary ligands (Ru-trz, Ir(I)-trz) or overall decomposition (Rh(I) and Os-trz), though no sign of trz–metal bond cleavage was detected (Fig. S26–S32, ESI†). These data indicate a stable M–Ctrz bond in all complexes but the silver system. Notably, the overall complex stability is influenced by the nature of the NHC and the metal and is higher for Ag complexes bound to trz than to imi, while the inverse was noted for Ru and Ir complexes. Ancillary ligand dissociation in these complexes is slower in the imi analogues.
As noted for complexes bearing imi ligands, stability differences in the M-trz series were only observed at elevated temperatures. At 120 °C, the same four reactivity patterns were observed (Scheme 3; Table S6 and Fig. S44–S52, ESI†). The gold(I) complex Au-trz was completely stable and no modification was observed over 24 h (Fig. 3). Complexes with PdII and IrI decomposed without formation of thione nor triazolium salt, as observed for their imi analogues but at different rates. The degradation is slow for Pd-trz (5% within 24 h) and fast for Ir(I)-trz (100% in 30 min; Fig. S45 and S46, ESI†). Trz complexes of OsII, RuII and RhI revealed some thione formation after modification of the initial complex. As observed for their imi analogues, loss of ancillary ligands was already observed after 30 min and almost full degradation after 24 h, with only 22–50% thione formed. Loss of cymene was observed in Os-trz after 30 min (20%) and almost full degradation after 24 h (28% thione, Fig. S49, ESI†). Similarly, Ru-trz slowly lost p-cymene (22% after 30 min) and fully decomposed after 24 h (50% of thione), while Rh(I)-trz lost COD much faster (full decomposition after 30 min and 22% thione after 24 h, Fig. S47 and S48, ESI†). Finally, complex Rh(III)-trz produced some quantities of triazolium salt and over time also thione (full degradation, 72% [trz-H]+ and 23% trz = S after 6 h, Fig. S51, ESI†).‡ Complex Ir(III)-trz decomposed slower than the Rh(III) analogue, with formation of thione but no triazolium salt (62% decomposition and 57% trz = S after 6 h, Fig. S50, ESI†).
 | ||
| Scheme 3 NHC dissociation pathways from M-trz complexes leading to thione trz = S or triazolium salt [trz-H]+ and complex degradation via ancillary ligand dissociation. | ||
 | ||
| Fig. 3 Amount of thione, starting complex, imidazolium salt, and degradation products after 24 h reaction at 120 °C, for M-trz complexes. Some triazolium salt [trz-H]+ was observed transiently from Rh(III)-trz after 30 min. | ||
The nature of the NHC had distinct effects on the NHC–metal bond stability depending on the coordinated metal. The trz ligand is more labile in group 8 metal complexes (Ru, Os) while higher stability was observed for group 11 metal complexes (Ag; Fig. S2, ESI†). The increased stability of trz bonds to d8/d10 metals (Rh(I), Ir(I), Ag, Cu) as opposed to d6 metals (Ru, Os) is remarkable since trz ligands were shown to be stronger donors than imi,29 which would suggest tighter bonding with less electron-rich metal centres. Obviously, the NHC–metal bond stability may significantly deviate from the trends established here, e.g., by wingtip group variations, by modified conditions such as different polarity of the solvent, by variation of the ancillary ligands and their electronic and steric properties, or by the presence of additives such as base or acid as used e.g. under catalytic conditions. For example, Ir(III)-trz, which is catalytically active in transfer hydrogenation at 80 °C in basic iPrOH does not show any degradation at this temperature in dichlorobenzene (Fig. S58 and S59, ESI†). Nonetheless, these results indicate that the preservation of the NHC–metal bond should not be taken as granted and mechanistic proposals should be tested under relevant conditions for NHC ligand dissociation rather than assuming by default a reliable bonding of the NHC ligand to the metal centre throughout a catalytic cycle, as often assumed due to the relatively high covalent character of the M–NHC bond. These conclusions align well with earlier studies that have revealed M–NHC bond dissociation as the crucial step for catalyst activation.23 Limited M–NHC stability may also be a critical factor for the recently established anticancer activity of some NHC metal complexes.30
In summary, this work provides insights into the intrinsic stability of the NHC–metal bond of both Arduengo-type imidazolylidenes as well as mesoionic triazolylidenes bound to metal centres of catalytic relevance (Fig. S58, ESI†). Under the specific conditions and in the absence of any auxiliary or substrate, several complexes including IrIII, RhIII, and NiII showed kinetic lability of the carbene metal bond, with slightly better stability of triazolylidenes than imidazolylidenes. This intrinsic kinetic lability of the M–NHC bond has obvious ramifications in catalysis and needs to be considered in catalyst design. Moreover, caution is required when assuming a robust M–NHC bond in mechanistic proposals. This lability of the NHC–metal bond can be effectively mitigated, for example, by installing appropriate chelating groups on the NHC ligand.
We thank the Swiss National Science Foundation (200020-182633) for generous financial support and the Marie Sklodowska-Curie Action for a fellowship to N. S. (grant 796762).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. J. Arduengo and G. Bertrand, Chem. Rev., 2009, 109, 3209–3210 CrossRef CAS PubMed.
- L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC.
- E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- C. S. J. Cazin, N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, Springer, Netherlands, Dordrecht, 2011 Search PubMed.
- N-Heterocyclic Carbenes, ed. S. Diez-Gonzalez, The Royal Society of Chemistry, 2017 Search PubMed.
- C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801–5809 CrossRef CAS.
- H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS.
- L. Busetto, M. C. Cassani, C. Femoni, A. Macchioni, R. Mazzoni and D. Zuccaccia, J. Organomet. Chem., 2008, 693, 2579–2591 CrossRef CAS.
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed., 2003, 36, 2162–2187 CrossRef.
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- S. Kuwata and F. E. Hahn, Chem. Rev., 2018, 118, 9642–9677 CrossRef CAS PubMed.
- F. E. Hahn, C. Radloff, T. Pape and A. Hepp, Chem. – Eur. J., 2008, 14, 10900–10904 CrossRef CAS PubMed.
- A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572–4573 CrossRef CAS PubMed.
- (a) G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed; (b) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- K. J. Cavell and D. S. McGuiness, Coord. Chem. Rev., 2004, 248, 671–681 CrossRef CAS.
- (a) D. Canseco-Gonzalez, A. Gniewek, M. Szulmanowicz, H. Müller-Bunz, A. Trceziak and M. Albrecht, Chem. – Eur. J., 2012, 18, 6055–6062 CrossRef CAS PubMed; (b) D. Canseco-Gonzalez, A. Petronilho, H. Müller-Bunz, K. Ohmatsu, T. Ooi and M. Albrecht, J. Am. Chem. Soc., 2013, 135, 13193–13203 CrossRef CAS PubMed; (c) V. M. Chernyshev, O. V. Khazipov, M. A. Shevchenko, A. Y. Chernenko, A. V. Astakhov, D. B. Eremin, D. V. Pasyukov, A. S. Kashin and V. P. Ananikov, Chem. Sci., 2018, 9, 5564–5577 RSC; (d) V. M. Chernyshev, E. A. Denisova, D. B. Eremin and V. P. Ananikov, Chem. Sci., 2020, 11, 6957–6977 RSC.
- T. K. Das and A. T. Biju, Eur. J. Org. Chem., 2017, 4500–4506 CrossRef CAS.
- J. Zhang, M. Franz, E. Hübner and A. Schmidt, Tetrahedron, 2016, 72, 525–531 CrossRef CAS.
- G. Tan and H. Zhu, Inorg. Chem., 2011, 50, 6979–6986 CrossRef CAS PubMed.
- E. Caytan and S. Roland, Organometallics, 2014, 33, 2115–2118 CrossRef CAS.
- H.-L. Su, L. M. Pérez, S.-J. Lee, J. H. Reibenspies, H. S. Bazzi and D. E. Bergbreiter, Organometallics, 2012, 31, 4063–4071 CrossRef CAS.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- (a) D. Truong, M. P. Sullivan, K. K. H. Tong, T. R. Steel, A. Prause, J. H. Lovett, J. W. Andersen, S. M. F. Jamieson, H. H. Harris, I. Ott, C. M. Weekley, K. Hummitzsch, T. Söhnel, M. Hanif, N. Metzler-Nolte, D. C. Goldstone and C. G. Hartinger, Inorg. Chem., 2020, 59, 3281–3289 CrossRef CAS PubMed; (b) I. M. Daubit, M. P. Sullivan, M. John, D. C. Goldstone, C. G. Hartinger and N. Metzler-Nolte, Inorg. Chem., 2020, 59, 17191–17199 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and NMR spectra for all new complexes, tables and NMR spectra of all stability tests. See DOI: 10.1039/d1cc02740a |
| ‡ As observed with [imi-H]+, the triazolium salt [trz-H]+ is gradually converted into the thione trz = S, with a higher yield than the imi system (71% vs. 30% conversion in 24 h; Fig. S52, ESI†). |
| This journal is © The Royal Society of Chemistry 2021 |