
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccessing novel fluorinated heterocycles with the hypervalent fluoroiodane reagent by solution and mechanochemical synthesis†
William
Riley
a,
Andrew C.
Jones
 b,
Kuldip
Singh
a,
Duncan L.
Browne
*c and
Alison M.
Stuart
*a
b,
Kuldip
Singh
a,
Duncan L.
Browne
*c and
Alison M.
Stuart
*a
aSchool of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: Alison.Stuart@le.ac.uk
bCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
cSchool of Pharmacy, UCL, London, WC1N 1AX, UK. E-mail: Duncan.Browne@ucl.ac.uk
First published on 14th June 2021
Abstract
A new and efficient strategy for the rapid formation of novel fluorinated tetrahydropyridazines and dihydrooxazines has been developed by fluorocyclisation of β,γ-unsaturated hydrazones and oximes with the fluoroiodane reagent. Mechanochemical synthesis delivered fluorinated tetrahydropyridazines in similar excellent yields to conventional solution synthesis, whereas fluorinated dihydrooxazines were prepared in much better yields by ball-milling.
Over 85% of small molecule pharmaceuticals approved by the FDA in 2019 contained a nitrogen-based heterocycle and over a quarter of all FDA approved drug molecules on the market contain at least one fluorine atom.1a The prominence of heterocycles comes from their activity in vivo, where, specifically their structure provides rigidity to molecular shape and enables effective binding. The pharmaceutical industry is particularly interested in heterocycles with a high sp3 content1b and the C–F bond has a great influence on the pharmacokinetic properties of biologically active molecules.1c
Fluorinated N-heterocycles can be accessed in a single step by intramolecular fluorocyclisations of unsaturated substrates containing an internal nucleophile. The fluorocyclisations of β,γ-unsaturated hydrazones with Selectfluor produced the 5-membered, fluorinated dihydropyrazoles (Scheme 1A).2 In one report, fluorinated tetrahydropyridazines were formed along with the dihydropyrazoles when R was an aryl group.2 Similarly, the fluorocyclisations of β,γ-unsaturated oximes formed the 5-membered, fluorinated dihydroisoxazoles using either Selectfluor,3a,b or hypervalent iodine reagent, PhI(OPiv)2, with HF-pyridine.3c The synthesis of the 6-membered, fluorinated dihydrooxazines has not been reported (Scheme 1B).
 | ||
| Scheme 1 (A) Synthesis of fluorinated dihydropyrazoles. (B) Synthesis of fluorinated dihydroisoxazoles. (C) Synthesis of fluorinated tetrahydropyridazines and dihydrooxazines. | ||
Since its inception, the hypervalent iodine(III) reagent, fluoroiodane 1 (Scheme 1C), has emerged as an excellent fluorinating reagent with a wide range of applications.4,5 In many cases, fluoroiodane 1 exhibits differing reactivity to that observed with fluoraza reagents such as Selectfluor. We recently employed fluoroiodane 1 in the synthesis of fluorinated lactones using either AgBF46a or hexafluoroisopropanol (HFIP)6b to activate the fluoroiodane reagent. Looking to further explore the utility of this reagent in fluorocyclisations, we investigated the synthesis of fluorinated tetrahydropyridazines and dihydrooxazines. We also applied ball-milling to the fluorocyclisations since mechanochemical synthesis can minimise solvent requirement and in many cases, can provide enhanced and altered reactivity.7 Ball-milling has shown promise in bromocyclisations8 and in fluorination reactions, where Selectfluor can selectively mono- or di-fluorinate 1,3-dicarbonyl compounds,9 fluorinate pyrazolone rings,10a and be used in asymmetric organocatalysis.10b Herein, we report the regioselective synthesis of novel fluorinated 6-membered heterocycles, tetrahydropyridazines and dihydrooxazines, alongside the use of mechanochemistry to minimise halogenated solvents and to improve the yields in the preparation of fluorinated dihydrooxazines.
We began our studies by investigating the fluorocyclisation of hydrazone 2a. To our delight, 2a was fluorocyclised at room temperature in 4 hours using fluoroiodane 1 (1.5 equiv.) with AgBF4 (1 equiv.) in CH2Cl2 to produce the desired fluorinated tetrahydropyridazine 3a in 91% yield (Table 1, entry 1). The reaction time could be reduced to just 15 minutes with minimal decrease in yield (entry 3). Alternatively, the amount of AgBF4 could be decreased to 0.2 equivalents in a 1 hour reaction to form the product in 86% yield (entry 4). On further investigation of solvent and temperature (entries 5–10), a set of metal-free conditions were identified using HFIP as the solvent and activator of fluoroiodane 1 giving 3a in 93% yield after only 15 minutes (entry 10). Notably, whilst use of acetonitrile as solvent did furnish the desired fluorinated product 3a, Ritter product 4 was also formed in 23% yield suggesting that a tertiary carbocation is a key intermediate in the fluorocyclisation. In entries 7 and 8 where product yield was lower at 40 °C, elimination byproduct 5 was also observed in 10–15% yield.

|
Using mechanochemistry the unsaturated hydrazones could efficiently undergo fluorocyclisations with fluoroiodane 1 (1.5 equiv.) and just 2 equivalents of HFIP (an 8 fold reduction in the amount of HFIP required, ESI,† Table S3) to provide an alternative, solvent minimised method. For this protocol, a 10 mL stainless steel milling jar was used with a 2.5 g stainless steel milling ball, and the reaction mixture was milled at 30 Hz for 15 minutes.
With optimal conditions for both a solution and ball-milling method in hand, application to a range of unsaturated hydrazones was explored (Scheme 2). The reaction was tolerant for a series of aryl substituted hydrazones (Scheme 2, 3a–h) with examples of both electron-donating (3e) and electron-with- drawing aryl groups (3b). Although synthesis of the thiophene substituted hydrazone proceeded efficiently (3h), the aromatic byproduct, 5-methyl-3-thiophen-2-yl pyridazine, was also formed in 16% yield. The fluorocyclisation proved successful on alkyl substituted hydrazones (3i–k) including benzyl and the more hindered isopropyl hydrazones. Bis-tetrahydropyridazine (3l) was prepared by a double fluorocyclisation. In all cases, metal-free protocols could be employed and delivered similar yields in comparison to reactions containing AgBF4. The mechanochemical approach was applied to a small range of substrates (3b–e, 3k and 3l) and provided comparable excellent yields to those observed in solution demonstrating an efficient, solvent minimised method. Further studies revealed that catalytic amounts of HFIP (0.1 equiv.) activated fluoroiodane 1 in CH2Cl2 and delivered fluorinated tetrahydropyridazine 3a in 90% yield (Scheme S1, ESI†).
 | ||
| Scheme 2 Scope of 5-fluorinated tetrahydropyridazine synthesis by solution and mechanochemical synthesis. | ||
Initial results with β,γ-unsaturated oximes demonstrated that they could undergo fluorocyclisation to form dihydro-oxazines. This class of fluorinated heterocycle has not been reported previously. After optimisation for traditional solution conditions (ESI,† Table S4), dihydrooxazine 7a could be formed in 46% yield after 15 minutes with 1 equivalent of AgBF4 (Table 2A). Notably, dihydroisoxazole 8, intercepted by the benzyl-alcohol backbone of the fluoroiodane reagent, was also isolated as a side product in 15% yield. In an attempt to improve yield and selectivity towards the desired product, attention was turned to mechanochemical techniques commencing with the conditions used for hydrazone fluorocyclisations (Scheme 2). Fluorocyclisation of unsaturated oxime 6b using fluoroiodane 1 proceeded to give 36% of the desired product with 2 equivalents of HFIP (Table 2, entry 1). Increased loading of HFIP improved the yield of dihydrooxazine 7b, but also increased the amount of unwanted dihydroisoxazole 9 (entry 2). A small amount of oxazine 10, resulting from elimination, was also observed. Switching to AgBF4 showed decreased activity initially (entry 3). However, using AgBF4 with 5 equivalents of CH2Cl2 provided a dramatic improvement in the yield of dihydrooxazine 7b (80% isolated yield), and reduced the formation of side products 9 and 10 to 1% (entry 5). Further studies on the two additives and reaction time revealed that 2 equivalents of AgBF4 and 5 equivalents of CH2Cl2 in a 1 hour reaction was optimal (entries 6–10). In the absence of HFIP or AgBF4, the reaction showed no activity and returned quantitative amounts of oxime 6b (entry 11).
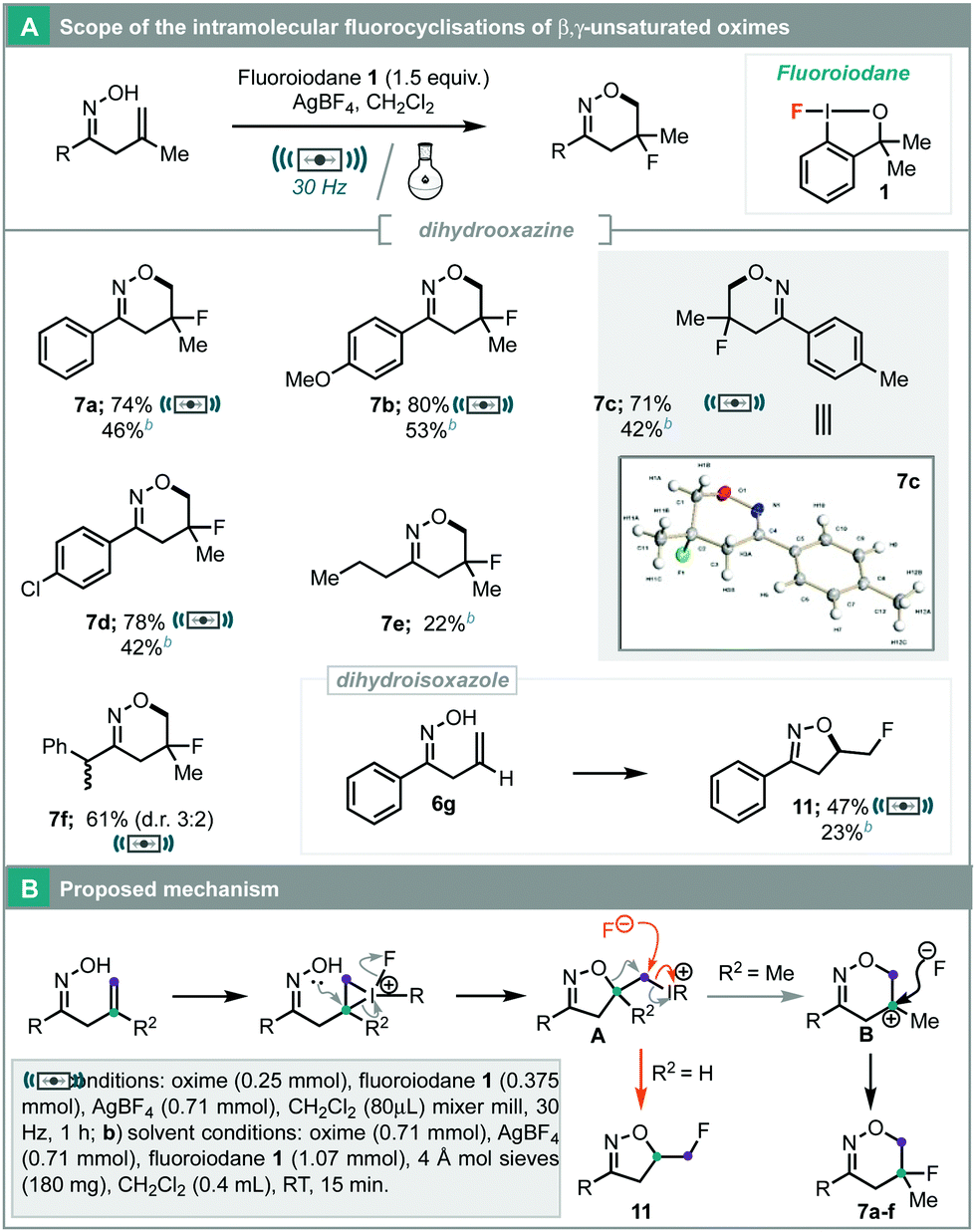 | ||
| Scheme 3 (A) Scope of 5-fluorodihydrooxazine synthesis. (B) Proposed mechanism. | ||

|
A range of β,γ-unsaturated oximes was examined and under traditional solution conditions, aryl unsaturated oximes could undergo fluorocyclisations in moderate yield (Scheme 3A, 7a–7d, 42–53%). However, applying mechanochemical conditions to these substrates greatly improved the yields delivering products 7a–7d in 71–80% yield. Alkyl oximes could also be tolerated (Scheme 3A, 7e–7f), but oxime 6e gave a lower yield as it contained a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of (Z)- and (E)-isomers and only the (Z)-isomer fluorocyclised. Unsaturated oxime 6g with a mono-substituted alkene selectively formed dihydroisoxazole 11, as reported by Wang using PhI(OPiv)2 and HF·pyridine.3c This result supports our proposed mechanism (Scheme 3B) where there is initial formation of a 5-membered heterocycle A, followed by ring expansion via tertiary carbocation B, to afford the 6-membered fluorinated heterocycle. However, for mono-substituted unsaturated oximes, formation of a secondary carbocation is not favoured and instead attack of fluoride at the methylene site forms the 5-membered product. Good evidence that tertiary carbocation B is a key intermediate was provided by the formation of Ritter product 4 in the fluorocyclisation of 2a in acetonitrile (Table 1, entry 6). Hara has also reported a similar ring expansion in the fluorocyclisation of 1-decen-5-ol with iodotoluene difluoride.11,12
:1 mixture of (Z)- and (E)-isomers and only the (Z)-isomer fluorocyclised. Unsaturated oxime 6g with a mono-substituted alkene selectively formed dihydroisoxazole 11, as reported by Wang using PhI(OPiv)2 and HF·pyridine.3c This result supports our proposed mechanism (Scheme 3B) where there is initial formation of a 5-membered heterocycle A, followed by ring expansion via tertiary carbocation B, to afford the 6-membered fluorinated heterocycle. However, for mono-substituted unsaturated oximes, formation of a secondary carbocation is not favoured and instead attack of fluoride at the methylene site forms the 5-membered product. Good evidence that tertiary carbocation B is a key intermediate was provided by the formation of Ritter product 4 in the fluorocyclisation of 2a in acetonitrile (Table 1, entry 6). Hara has also reported a similar ring expansion in the fluorocyclisation of 1-decen-5-ol with iodotoluene difluoride.11,12
It is intriguing that a rearrangement, driven by carbocation stability, still occurs under solvent minimised conditions in a ball-mill. On a similar thread, we also explored ball-milling techniques in the already established fluoroiodane mediated fluorolactonisation to furnish a range of γ- and δ-lactones 13a–f (Scheme 4).6b These products can only be obtained from a reaction mechanism involving a fluorocyclisation and an aryl migration (Scheme S2, ESI†).6a It is remarkable that the aryl migration occurred in the ball-mill with a 6 fold reduction in the volume of HFIP. To demonstrate this further, treatment of unsaturated carboxylic acid 12f with Selectfluor under ball-milling conditions for 1 hour at 30 Hz furnished fluorolactonised product 14 without an aryl migration, and so, the product could be switched by merely changing Selectfluor for fluoroiodane.
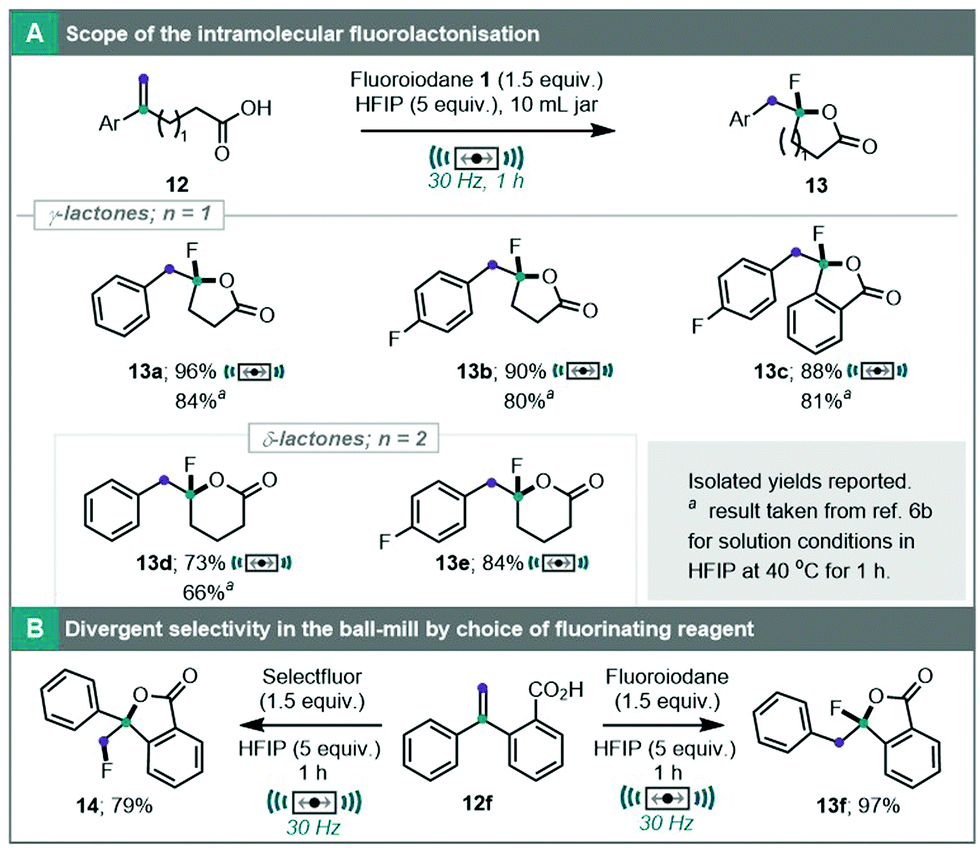 | ||
| Scheme 4 (A) Scope of ball-milled intramolecular fluorolactonisation. (B) Switching of cyclisation selectivity by choice of fluorinating reagent. | ||
In summary, we have developed efficient protocols for accessing two new classes of fluorinated heterocycles using fluoroiodane reagent 1. Alternative selectivity to that displayed by electrophilic fluorinating reagents (e.g. Selectfluor) is clearly demonstrated and thus, this study shows that fluoroiodane 1 is a complementary reagent for diversifying fluorination methodology. Mechanochemistry has provided a dramatic improvement in the yield and selectivity to fluorinated dihydrooxazines, and in so doing has highlighted that mechanistically complex reactions can be readily achieved in a ball-mill.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. G. de la Torre and F. Albericio, Molecules, 2020, 25, 745 CrossRef CAS PubMed; (b) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- J. Zhao, M. Jiang and J.-T. Liu, Org. Chem. Front., 2018, 5, 1155 RSC.
- (a) Y.-Y. Liu, J. Yang, R.-J. Song and J.-H. Li, Adv. Synth. Catal., 2014, 356, 2913 CrossRef CAS; (b) J. Zhao, M. Jiang and J.-T. Liu, Adv. Synth. Catal., 2017, 359, 1626 CrossRef CAS; (c) W. Kong, Q. Guo, Z. Xu, G. Wang, X. Jiang and R. Wang, Org. Lett., 2015, 17, 3686 CrossRef CAS PubMed.
- (a) C. Y. Legault and J. Prévost, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2012, 68, o1238 CrossRef CAS PubMed; (b) V. Matoušek, E. Pietrasiak, R. Schwenk and A. Togni, J. Org. Chem., 2013, 78, 6763 CrossRef PubMed; (c) G. C. Geary, E. G. Hope, K. Singh and A. M. Stuart, Chem. Commun., 2013, 49, 9263 RSC.
- (a) N. O. Ilchenko, B. O. A. Tasch and K. J. Szabó, Angew. Chem., Int. Ed., 2014, 53, 12897 CrossRef CAS PubMed; (b) G. C. Geary, E. G. Hope, K. Singh and A. M. Stuart, RSC Adv., 2015, 5, 16501 RSC; (c) W. Yuan and K. J. Szabó, Angew. Chem., Int. Ed., 2015, 54, 8533 CrossRef CAS PubMed; (d) A. Ulmer, C. Brunner, A. M. Arnold, A. Pöthig and T. Gulder, Chem. – Eur. J., 2016, 22, 3660 CrossRef CAS PubMed; (e) W. Yuan, L. Eriksson and K. J. Szabó, Angew. Chem., Int. Ed., 2016, 55, 8410 CrossRef CAS PubMed; (f) N. O. Ilchenko, M. Hedberg and K. J. Szabó, Chem. Sci., 2017, 8, 1056 RSC; (g) B. Xing, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2018, 57, 9896 CrossRef CAS PubMed; (h) A. M. H. Abudken, E. G. Hope, K. Singh and A. M. Stuart, Org. Biomol. Chem., 2020, 18, 6140 RSC; (i) S. Yang, S. Shi, Y. Chen and Z. Ding, J. Org. Chem., 2021, 86 DOI:10.1021/acs.joc.1c00159.
- (a) G. C. Geary, E. G. Hope and A. M. Stuart, Angew. Chem., Int. Ed., 2015, 54, 14911 CrossRef CAS PubMed; (b) H. K. Minhas, W. Riley, A. M. Stuart and M. Urbonaite, Org. Biomol. Chem., 2018, 16, 7170 RSC.
- (a) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080 RSC; (b) G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC; (c) A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317 RSC; (d) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018 CrossRef PubMed; (e) J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007 CrossRef PubMed; (f) A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344 CrossRef CAS; (g) J. Andersen and J. Mack, Green Chem., 2018, 20, 1435 RSC; (h) S. L. James, et al. , Chem. Soc. Rev., 2012, 41, 413 RSC; (i) M. Leonardi, M. Villacampa and J. C. Menéndez, Chem. Sci., 2018, 9, 2042 RSC; (j) T.-X. Métro, J. Martinez and F. Lamaty, ACS Sustainable Chem. Eng., 2017, 5, 9599 CrossRef.
- J. Wong and Y.-Y. Yeung, RSC Adv., 2021, 11, 13564 RSC.
- (a) J. L. Howard, Y. Sagatov, L. Repusseau, C. Schotten and D. L. Browne, Green Chem., 2017, 19, 2798 RSC; (b) J. L. Howard, Y. Sagatov and D. L. Browne, Tetrahedron, 2018, 74, 3118 CrossRef CAS; (c) Y. Wang, H. Wang, Y. Jiang, C. Zhang, J. Shao and D. Xu, Green Chem., 2017, 19, 1674 RSC.
- (a) J. L. Howard, W. Nicholson, Y. Sagatov and D. L. Browne, Beilstein J. Org. Chem., 2017, 13, 1950 CrossRef CAS PubMed; (b) D. Krištofíková, M. Mečiarová, E. Rakovský and R. Šebesta, ACS Sustainable Chem. Eng., 2020, 8, 14417 CrossRef.
- M. Sawaguchi, S. Hara, T. Fukuhara and N. Yoneda, J. Fluorine Chem., 2000, 104, 277 CrossRef CAS.
- For ring expansion of cyclic ethers with p-iodotoluene difluoride see: T. Inagaki, Y. Nakamura, M. Sawaguchi, N. Yoneda, S. Ayuba and S. Hara, Tetrahedron Lett., 2003, 44, 4117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR spectra. CCDC 2083051–2083056. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc02587b |
| This journal is © The Royal Society of Chemistry 2021 |