
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A computationally designed β-amino acid-containing miniprotein†
Magdalena
Bejger‡
 a,
Paulina
Fortuna‡
bc,
Magda
Drewniak-Świtalska
b,
Jacek
Plewka
d,
Wojciech
Rypniewski
a and
Łukasz
Berlicki
*b
a,
Paulina
Fortuna‡
bc,
Magda
Drewniak-Świtalska
b,
Jacek
Plewka
d,
Wojciech
Rypniewski
a and
Łukasz
Berlicki
*b
aInstitute of Bioorganic Chemistry, Polish Academy of Sciences, Noskowskiego 12/14, Poznań 61-704, Poland
bDepartment of Bioorganic Chemistry Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, Wrocław 50-370, Poland. E-mail: lukasz.berlicki@pwr.edu.pl
cDepartment of Medical Biochemistry, Wrocław Medical University, Pausteura 1, Wroclaw 50-368, Poland
dFaculty of Chemistry, Jagiellonian Univeristy, Gronostajowa 2, Kraków 30-387, Poland
First published on 12th May 2021
Abstract
A new miniprotein built from three helices, including one structure based on the ααβαααβ sequence pattern was developed. Its crystal structure revealed a compact conformation with a well-packed hydrophobic core of unprecedented structure. The miniprotein formed dimers that were stabilized by the interaction of their hydrophobic surfaces.
Since the discovery of the unexpected structural features of β-peptides1,2 published in the 1990s, the field of β-amino acid-containing peptides has grown immensely. The acquired knowledge concerns both a wide range of diverse structural studies3 and numerous applications.4 However, the vast majority of structural studies are focused on secondary structures, particularly helices. A surprising diversity of helical conformations can be found, which depend on the β-amino acid, sequence pattern and the stereochemistry used.5 Various helix parameters, including diameter, pitch, side chain spatial distribution and handedness, can be achieved. The propensity of individual α/β-peptides to form a helical structure is described using the so-called ‘stereochemical patterning’ theory.6 If the amino acid units show a tendency to induce dihedral angles of the same sign that are flanking an amide bond, it is expected that a helix will be formed. In particular, structurally constrained, cycloalkane-based β-amino acid residues have a significant impact on the folding propensities of peptides. Amino acids incorporating cycloalkanes containing 3 to 6 atoms have been used for the construction of conformationally stable peptides.7 Peptides with various stereochemistries as well as sequence patterns (αββ, αβ, ααβ, ααββ, etc.) have been shown to fold.8 It is worth noting that the unique structural features of α/β-peptides provide new opportunities for numerous applications. Biological activities9 including antimicrobial10 and antiangiogenic properties11 and the inhibition of protein–protein interactions12 and agonism/antagonism of G protein coupled receptors (GPCR),13 have been found. Moreover, possibilities for the construction of efficient α/β-peptide-based catalysts have also been revealed.14
Although studies on α/β-peptides adopting secondary structures have led to a large number of successful research studies, analysis of more extended structures is very limited and no methodology for the de novo design of such structures has been published until now.15 Published studies provide insights into the effects of substitution of known miniproteins (i.e., peptides of 20–50 amino acid residues that form stable tertiary structures in solution) by various β-amino acids. Studies on the analogs of the villin headpiece subdomain (VHP),16 the B domain of protein G (GB1),17 betabellin-14,18 Trp-cage, and FSD19 indicated that mutations of certain residues of these miniproteins could provide stable analogs; however, in most cases, such substitutions led to a decrease in conformational stability in comparison to the stability of their native counterparts. A higher conformational stability could be achieved for substitutions at terminal positions of the helical structures. The use of cycloalkane-based constrained β-amino acids usually results in analogs with a lower enthalpy but a higher entropy for the folding process. The enthalpy effect is attributed to a less favorable net of intramolecular interactions related to the altered conformation of the molecule. On the other hand, a beneficial increase in entropy is related to the preorganization of the molecule towards a properly folded conformation.
The design of protein-like structures remains a great challenge in modern chemistry and has not yet been achieved for β-amino acid-containing miniproteins. In this paper, we explore the possibility of the construction of a miniprotein composed of three helical fragments: two α-helices and one helix with an altered conformation by the application of trans-(1S,2S)-2-aminocyclopentanecarboxylic acid (trans-ACPC). Helices were arranged in a similar manner to those in the native miniprotein, the VHP20 (Fig. 1), but the construction of the hydrophobic core and intramolecular interactions responsible for its conformational stability were different. As expected, sequence similarity between VHP and peptides 1–5 is low (23–31%), while that between peptides 1–5 is high (66–80%, Fig. S1, ESI†). The similar size and secondary structure composition will allow a comparison of the native and artificially constructed miniproteins. Computer-aided design of β-amino acid-containing miniproteins was based on the FastDesign protocol and was implemented using the Rosetta software package.21 Two short α-helical fragments joined by a glycine residue were attached to a helical fragment constructed using the ααβαααβ sequence pattern with trans-ACPC. The conformation of this β-amino acid-modified helix was based on the published crystal structure of a helical fragment with the same pattern.22 The side chains were designed to form a well-packed hydrophobic core. Among numerous modeled sequences, five miniproteins with the highest estimated stability were chosen for synthesis (peptides 1–5, Fig. 1 and Table S1, ESI†).
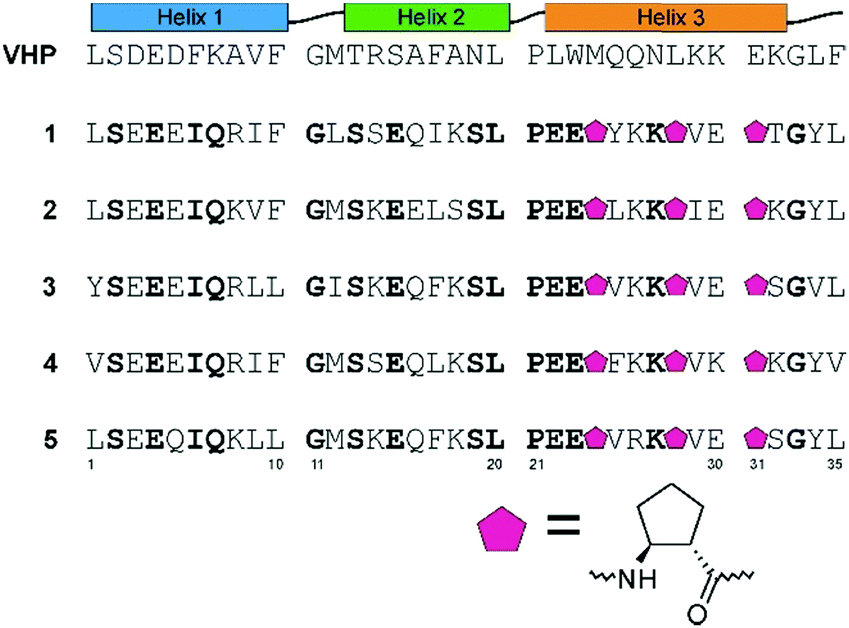 | ||
| Fig. 1 The design principles, sequences of villin (VHP) and the studied peptides (1–5). Amino acid residues shown in bold are conserved among peptides 1–5. All the peptides are amidated at the C-terminus; in all cases, methionine was exchanged to norleucine. | ||
All the indicated peptides were successfully obtained using microwave-assisted automated solid phase peptide synthesis by applying Fmoc chemistry (Table S2, ESI†). CD spectra measured for peptides dissolved in phosphate buffer at pH 7.0 indicated a high content of helical structures for compounds 1–3 and 5 (Fig. 2). The CD spectrum of peptide 4 had a minimum at approximately 202 nm, which corresponds to a disordered structure in solution. It is worth noting that the CD spectra of synthesized α/β-peptides differ significantly from a typical CD spectrum observed for α-helix-containing proteins (e.g., VHP,16 two deep minima at 208 and 222 nm). These changes in the CD spectra are related to the presence of a helix whose conformation is significantly altered by the presence of constrained β-amino acid residues.
 | ||
| Fig. 2 CD spectra of the studied peptides 1–5 dissolved in phosphate buffer at pH 7.0 at 293 K. MRE – mean residue ellipticity. | ||
Subsequently, the evaluation of the conformational stability of newly designed miniproteins was performed by temperature scans monitored by CD spectroscopy (Fig. 3). Consistent with the CD spectra, four of five new peptides (compounds 1–3 and 5) showed a sigmoidal shape curve of temperature dependence, which is typical of cooperatively folding proteins. The estimated melting points of these oligomers were in the range of 39–69 °C. Peptides 1 and 3 showed values close to those observed for VHP (61.8 °C and 68.8 °C versus 69.1 °C16), indicating that the design process delivered well-folded peptides.
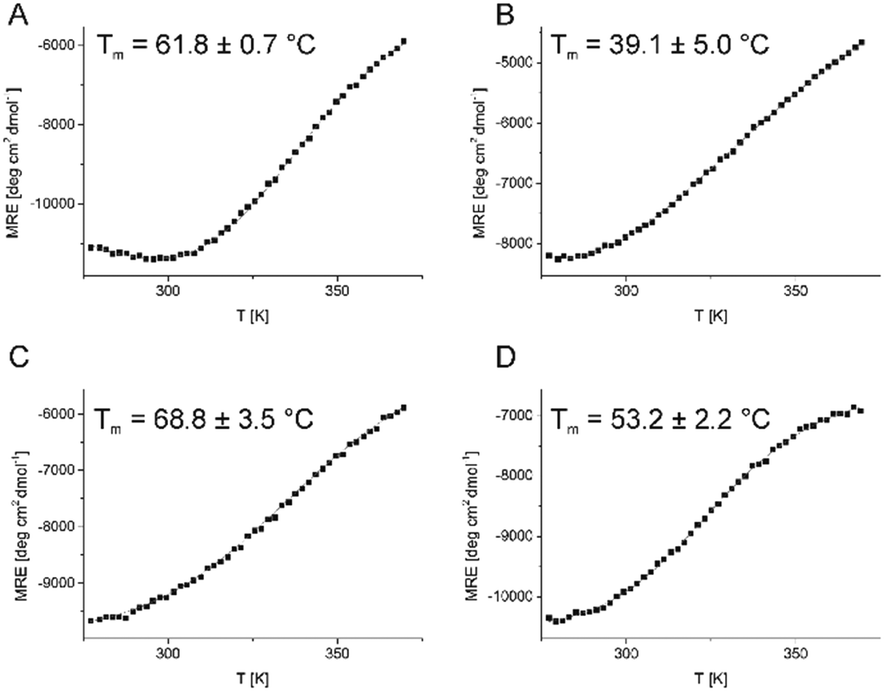 | ||
| Fig. 3 CD-monitored temperature dependence of peptides 1–3 and 5 (panels A–D, respectively) and their melting points (as insets). Experimental points (MRE at 222 nm) are denoted by black squares, while the fitted function is shown using a solid red line. | ||
More detailed studies were performed for peptide 1, for which the dependence of the CD signal on the temperature and the concentration of guanidine chloride (used as denaturant) was measured and fitted to the model, which was used to calculate the thermodynamic parameters of the unfolding process (Fig. S2, ESI†). The obtained ΔG0 value (at 293 K) is 0.48 kcal mol−1, and cp and m values are small and typical of miniproteins with a small hydrophobic core.
Extensive crystallization trials of the studied peptides resulted in triclinic crystals of peptide 1 that diffracted at a resolution of 1.1 Å. To phase the crystal structure using the anomalous dispersion method (Table S3, ESI†), a selenomethionine-substituted analog of peptide 1 (compound 1–Se) was prepared by exchanging the C-terminal leucine residue. Peptide 1–Se gave a monoclinic C2 crystal form diffracting at a resolution of 1.15 Å. The triclinic structure had four crystallographically independent molecules of peptide 1 in the asymmetric unit, while the monoclinic structure had two molecules in the asymmetric unit. All the molecules share the same fold, with minor local differences and varying amounts of disorder. Pairwise comparisons of individual chains show that most chains can be superposed with an RMSD below 0.5 Å. The connecting loop and termini are generally less ordered than the helices (Fig. S3, ESI†). The Se-derivatized structure 1–Se is less ordered than miniprotein 1 and shows a significant amount of alternative conformations, especially in the N-terminal helix, indicating that the helix can shift by approximately 1 Å without changing the overall fold.
The peptide 1 chain folds into a structure containing three helical fragments, including an α-helix (residues 3–10), a short 310 helix (residues 16–18) and a helix containing three β-amino acid residues (20–32), and is terminated by a short coil of three residues (Fig. 4 and Fig. S4, ESI†). The most prominent feature of the molecule is the helix containing the β-amino acids; it has a hydrogen bonding pattern of i + 4 − i that is characteristic of an α-helix and a similar number of residues per turn. The additional Cβ atoms present in the main chain of the β-amino acids are accommodated well within the α-helical geometry. The rings of the β-amino acids are on one side of the helix, making this side nonpolar. It is oriented towards a corresponding part of the other molecule that constitutes the dimer, while the other side of the helix is polar and faces the solvent. In both crystal structures (1 and 1–Se), the molecules dimerize in the same manner (Fig. 4B). The dimerization is evident upon visual inspection, but it was also identified unambiguously by the PDBePISA server.23 In general, polar residues form the exterior of the dimer, while nonpolar residues form the interior of the molecules and the dimerization interface. Another general feature of the structure is that while elements of the secondary structure are stabilized by hydrogen bonds, they are held together mainly by hydrophobic interactions. There are only four hydrogen bonds observed between the secondary structures (Gln7–Gly11, Gln7–Leu12, Gln16–Ser13 and Lys27–Phe10, Fig. S4A, ESI†). The dimerization interface is also hydrophobic in nature, and there are no hydrogen bonds between the protomers. It is also worth noting that although miniprotein 1 and VHP contain the same number of helices and sequences of the same length, their structures differ significantly in respect of both the construction of the hydrophobic core as well as the quaternary structure (Fig. 4A and D). It is noteworthy that the crystal structure of miniprotein 1 also differs from the initial design in respect of secondary structure relative positioning (Fig. 4A and C), which is most probably a result of the formation of conformation that is preferably creating a dimer interface.
 | ||
| Fig. 4 Crystal structure of peptide 1: monomer with residues forming a hydrophobic core shown as sticks (A) and dimer structure (B). Computer-aided design of peptide 1 (C) and structure of VHP (D) are shown for comparison. The main chain of all miniproteins is shown as a solid ribbon. trans-ACPC residues are shown as sticks with green carbon atoms. | ||
To disambiguate the oligomerization state of miniprotein 1 in solution, we performed a synchrotron radiation small angle X-ray scattering (sr-SAXS) experiment. The advantage of sr-SAXS over X-ray crystallography is that sr-SAXS experiments are measured in solution. Therefore, there are no artefacts arising from crystal packing, and the protein can be probed under native conditions. The mass of miniprotein 1 was estimated to be approximately 8 kDa (the ATSAS software), suggesting that it exists as a dimer in solution.24 Next, we extracted a dimer and a monomer from the high-resolution structure reported here and compared the putative scattering curves derived from them to the experimental curve. The curve representing a dimer fits the experimental data well (Fig. S5, ESI†) with a goodness-of-fit χ2 of 2.9, whereas both the tetramer and monomer presented poorer fits with χ2 values of 45.8 and 112.5, respectively. Moreover, using the Monte Carlo approach25 to tweak positions of the atoms in the high-resolution structure so that it resembles the SAXS signal more, the resulting dimer is slightly less compact with a radius of gyration of 1.33 nm compared to that of the native, 1.29 nm (Fig. S6, ESI†). This effect is also represented in the low molecular envelope calculated from the experimental sr-SAXS signal using DAMMIF, where there is “extra space” around the dimer (Fig. S5, ESI†).26
In summary, it has been shown experimentally that the α/β-hybrid miniprotein folds to an unprecedented well-defined three-dimensional structure. The miniproteins formed dimers, which were stable both in the solid phase and in solution. It is the first example of a β-amino acid containing miniprotein with a sequence that is not analogous to any previously known one. Moreover, it is also the first example of a β-amino acid containing-miniprotein that adopts quaternary structures. Therefore, it represents a significant breakthrough in peptide foldamers. The trans-ACPC substitution can not only rigidify the helical structure, which was shown previously, but also contribute to the formation of a hydrophobic core of a miniprotein and provide an interface for dimerization. In particular, the ααβαααβ sequence pattern, in which all cyclopentane-based amino acid residues are on the same side, is well suited for creating a zipper-like dimerization interface. In general, both the tertiary and quaternary structures are stabilized mainly by hydrophobic interactions, which are dependent on the structural complementarity of interacting partners, in contrast to secondary elements that largely dependent on hydrogen bonds. Therefore, all miniprotein organization levels, secondary, tertiary and quaternary, are favorably impacted by the presence of β-amino acid residues.
Taking into account numerous known types of helical structures containing β-amino acid residues and the experience provided by the presented study, the access to an entirely new world of protein mimetics is open. Due to their extended structure, the range of possible applications, in comparison to the already studied helix-based peptides, is significantly broadened and could allow demanding cases to be addressed, e.g. in the field of catalysis. Moreover, the combination of the beneficial features of β-amino acid-containing peptides (e.g. proteolytic stability) with the advantages of structurally extended protein-like molecules could provide scaffolds for the development of biologically active molecules, e.g. inhibitors of protein–protein interactions.
This work was financially supported by the National Science Centre, Poland (Grant no. DEC-2016/21/B/ST5/00269 to Ł. B.). The X-ray crystallographic data were collected on beamline P13, which is operated by EMBL Hamburg at the PETRA III storage ring. The SAXS experiments were performed on beamline BM29 at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). We are grateful to Dr Petra Pernot and Anton Popov at the ESRF for assistance in using beamline BM29.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015 RSC.
- (a) D. H. Appella, L. A. Christianson, D. A. Klein, D. R. Powell, X. Huang, J. J. Barchi Jr and S. H. Gellman, Nature, 1997, 387, 381 CrossRef CAS; (b) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS.
- (a) T. A. Martinek and F. Fülöp, Chem. Soc. Rev., 2012, 41, 687 RSC; (b) W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399 CrossRef CAS.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252 CrossRef CAS PubMed.
- L. M. Johnson and S. H. Gellman, Methods Enzymol., 2013, 523, 407 CAS.
- I. M. Mándity, E. Wéber, T. A. Martinek, G. Olajos, G. K. Tóth, E. Vass and F. Fülöp, Angew. Chem., Int. Ed., 2009, 48, 2171 CrossRef PubMed.
- (a) S. De Pol, C. Zorn, C. D. Klein, O. Zerbe and O. Reiser, Angew. Chem., Int. Ed., 2004, 43, 511 CrossRef CAS PubMed; (b) S. Izquierdo, F. Rua, A. Sbai, T. Parella, A. Alvarez-Larena, V. Branchadell and R. M. Ortuno, J. Org. Chem., 2005, 70, 7963 CrossRef CAS PubMed; (c) E. Torres, E. Gorrea, K. K. Burusco, E. Da Silva, P. Nolis, F. Ra, S. Boussert, I. Díez-Pérez, S. Dannenberg, S. Izquierdo, E. Giralt, C. Jaime, V. Branchadell and R. M. Ortuño, Org. Biomol. Chem., 2010, 8, 564 RSC; (d) S. Shin, M. Lee, A. I. Guzei, Y. Kee Kang and S. H. Choi, J. Am. Chem. Soc., 2016, 138, 13390 CrossRef CAS PubMed.
- (a) S. H. Choi, I. A. Guzei, L. C. Spencer and S. H. Gellman, J. Am. Chem. Soc., 2008, 130, 6544 CrossRef CAS PubMed; (b) S. H. Choi, I. A. Guzei, L. C. Spencer and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 2917 CrossRef CAS; (c) Ł. Berlicki, L. Pilsl, E. Wéber, I. M. Mándity, C. Cabrele, T. A. Martinek, F. Fülöp and O. Reiser, Angew. Chem., Int. Ed., 2012, 51, 2208 CrossRef PubMed; (d) M. Szefczyk, E. Węglarz-Tomczak, P. Fortuna, A. Krzysztoń, E. Rudzińska-Szostak and Ł. Berlicki, Angew. Chem., Int. Ed., 2017, 56, 2087 CrossRef CAS PubMed.
- C. Cabrele, T. A. Martinek, O. Reiser and Ł. Berlicki, J. Med. Chem., 2014, 57, 9718 CrossRef CAS PubMed.
- (a) M. A. Schmitt, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 417 CrossRef CAS PubMed; (b) M. van der Knaap, F. Basalan, H. C. van de Mei, H. J. Busscher, G. A. van der Marel, H. S. Overkleeft and M. Overhand, Chem. Biodiversity, 2012, 9, 2494 CrossRef CAS PubMed.
- Z. Hegedüs, E. Wéber, É. Kriston-Pál, I. Makra, Á. Czibula, É. Monostori and T. A. Martinek, J. Am. Chem. Soc., 2013, 135, 16578 CrossRef PubMed.
- (a) P. Fortuna, A. Twarda-Clapa, L. Skalniak, K. Ożga, T. A. Holak and Ł. Berlicki, Eur. J. Med. Chem., 2020, 208, 112814 CrossRef CAS PubMed; (b) H. S. Haase, K. J. Peterson-Kaufman, S. K. Lan Levengood, J. W. Checco, W. L. Murphy and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 7652 CrossRef CAS PubMed; (c) L. M. Johnson, D. E. Mortenson, H. G. Yun, W. S. Horne, T. J. Ketas, M. Lu, J. P. Moore and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 7317 CrossRef CAS PubMed.
- (a) Ł. Berlicki, M. Kaske, R. Gutierrez-Abad, G. Bernhardt, O. Illa, R. M. Ortuño, C. Cabrele, A. Buschauer and O. Reiser, J. Med. Chem., 2013, 56, 8422 CrossRef PubMed; (b) N. Koglin, C. Zorn, R. Beumer, C. Cabrele, C. Bubert, N. Sewald, O. Reiser and A. G. Beck-Sickinger, Angew. Chem., Int. Ed., 2003, 42, 202 CrossRef CAS PubMed.
- (a) M. M. Muller, M. A. Windsor, W. C. Pomerantz, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2009, 48, 922 CrossRef CAS; (b) M. Drewniak, E. Węglarz-Tomczak, K. Ożga, E. Rudzińska-Szostak, K. Macegoniuk, J. M. Tomczak, M. Bejger, W. Rypniewski and Ł. Berlicki, Bioorg. Chem., 2018, 81, 356 CrossRef CAS; (c) Z. C. Girvin, M. K. Andrews, X. Liu and S. H. Gellman, Science, 2019, 366, 1528 CrossRef CAS PubMed; (d) Z. C. Girvin and S. H. Gellman, J. Am. Chem. Soc., 2020, 142, 17211 CrossRef CAS PubMed.
- (a) K. L. George and W. S. Horne, Acc. Chem. Res., 2018, 51, 1220 CrossRef CAS PubMed; (b) R. David, R. Günther, L. Baumann, T. Lühmann, D. Seebach, H.-J. Hofmann and A. G. Beck-Sickinger, J. Am. Chem. Soc., 2008, 130(46), 15311 CrossRef CAS PubMed; (c) C. M. Lombardo, V. M. V. Kumar, C. Douat, F. Rosu, J.-L. Mergny, G. F. Salgado and G. Guichard, J. Am. Chem. Soc., 2019, 141(6), 2516 CrossRef CAS PubMed.
- D. F. Kreitler, D. E. Mortenson, K. T. Forest and S. H. Gellman, J. Am. Chem. Soc., 2016, 138, 6498 CrossRef CAS PubMed.
- (a) Z. E. Reinert and W. S. Horne, Chem. Sci., 2014, 5, 3325 RSC; (b) Z. E. Reinert, G. A. Lengyel and W. S. Horne, J. Am. Chem. Soc., 2013, 135, 12528 CrossRef CAS PubMed.
- G. Olajos, A. Hetényi, E. Wéber, L. J. Németh, Z. Szakonyi, F. Fülöp and T. A. Martinek, Chem. – Eur. J., 2015, 21, 6173 CrossRef CAS PubMed.
- M. Drewniak-Świtalska, B. Barycza, E. Rudzińska-Szostak, P. Morawiak and Ł. Berlicki, Org. Biomol. Chem., 2021, 19(19), 4272–4278 RSC.
- T. K. Chiu, J. Kubelka, R. Herbst-Irmer, W. A. Eaton, J. Hofrichter and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7517 CrossRef CAS PubMed.
- G. J. Rocklin, T. M. Chidyausiku, I. Goreshnik, A. Ford, S. Houliston, A. Lemak, L. Carter, R. Ravichandran, V. K. Mulligan, A. Chevalier, C. H. Arrowsmith and D. Baker, Science, 2017, 357, 168 CrossRef CAS PubMed.
- W. S. Horne, L. M. Johnson, T. J. Ketas, P. J. Klasse, M. Lu, J. P. Moore and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14751 CrossRef CAS PubMed.
- E. Krissinel and K. Henrick, J. Mol. Biol., 2007, 372, 774 CrossRef CAS PubMed.
- D. Franke, M. V. Petoukhov, P. V. Konarev, A. Panjkovich, A. Tuukkanen, H. D. T. Mertens, A. G. Kikhney, N. R. Hajizadeh, J. M. Franklin, C. M. Jeffries and D. I. Svergun, J. Appl. Crystallogr., 2017, 50, 1212 CrossRef CAS PubMed.
- M. V. Petoukhov and D. I. Svergun, Biophys. J., 2005, 89, 1237 CrossRef CAS PubMed.
- D. Franke and D. I. Svergun, J. Appl. Crystallogr., 2009, 42, 342 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc02192c |
| ‡ Authors with equal contributions. |
| This journal is © The Royal Society of Chemistry 2021 |