
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Higher MLCT lifetime of carbene iron(II) complexes by chelate ring expansion†
Thomas
Reuter
a,
Ayla
Kruse
b,
Roland
Schoch
 c,
Stefan
Lochbrunner
b,
Matthias
Bauer
c and
Katja
Heinze
*a
c,
Stefan
Lochbrunner
b,
Matthias
Bauer
c and
Katja
Heinze
*a
aDepartment of Chemistry, Johannes Gutenberg University of Mainz, Duesbergweg 10-14, Mainz 55128, Germany. E-mail: katja.heinze@uni-mainz.de
bInstitute for Physics and Department of Life, Light and Matter, University of Rostock, Rostock 18051, Germany
cFaculty of Science, Chemistry Department and Centre for Sustainable Systems Design, Paderborn University, Paderborn 33098, Germany
First published on 9th July 2021
Abstract
Combining strong σ-donating N-heterocyclic carbene ligands and π-accepting pyridine ligands with a high octahedricity in rigid iron(II) complexes increases the 3MLCT lifetime from 0.15 ps in the prototypical [Fe(tpy)2]2+ complex to 9.2 ps in [Fe(dpmi)2]2+12+. The tripodal CNN ligand dpmi (di(pyridine-2-yl)(3-methylimidazol-2-yl)methane) forms six-membered chelate rings with the iron(II) centre leading to close to 90° bite angles and enhanced iron-ligand orbital overlap.
Replacing expensive ruthenium(II) and iridium(III) complexes possessing long-lived metal-to-ligand charge transfer (MLCT) states in photochemical or photophysical applications with cheaper and more abundant alternatives is a highly important yet very challenging objective for future large-scale implementation.1–4 Iron(II) complexes, isoelectronic to RuII and IrIII have found tremendous interest for these purposes.5–8 However, due to a much smaller ligand field splitting,9 FeII complexes possess low-energy metal-centred states (3MC and 5MC) which provide efficient non-radiative relaxation pathways. Consequently, the MLCT photochemistry of iron(II) complexes is much less developed than for the classical ruthenium(II) or iridium(III) complexes.10,11 Concepts to prolong the lifetime of the potentially emissive 3MLCT state aim to increase the energy of the MC states and to decrease the energy of 3MLCT states.5–7
The latter has been targeted by using electron-poor π-accepting ligands12–15 while the energy of the MC states has been increased using a better metal–ligand orbital overlap in highly octahedral polypyridyl complexes with N–M–N angles close to 180°12–14 or strong σ-donating carbene ligands.6,7,15–20 The concept of large bite angles to separate 3MLCT from MC states had been very successful in the photophysical optimisation of ruthenium(II) sensitizers and phosphorescent emitters.21–25 In structurally related carbene pyridine iron(II) complexes with tridentate ligands forming five-membered chelate rings, the number of carbene donors dictates the 3MLCT lifetime.17 The highest lifetime (528 ps) of carbene iron(II) complexes has been achieved with six carbene donors in [Fe(btz)3]2+ (btz = 3,3’-dimethyl-1,1’-bis(p-tolyl))-4,4’-bis(1,2,3-triazol-5-ylidene).20 The effect of six-membered chelate rings to increase the metal ligand orbital overlap and consequently the energy of MC states of 3d metal complexes has been demonstrated with polypyridyl vanadium(III) and chromium(III) complexes26–28 as well as hexacarbene manganese(IV) and iron(III) complexes,29,30 while polypyridyl iron(II) complexes with high octahedricity still feature low MLCT lifetimes. The combination of four carbenes with six-membered chelate rings in iron(II) complexes with tridentate meridionally coordinating CNC ligands only provided low MLCT lifetimes of around 1 ps of the majority of the excited state population which has been ascribed to a higher flexibility of this CNC ligand.31
Here, we combine high octahedricity with π-accepting pyridine and σ-donating carbene ligands in a more rigid coordination environment to increase the 3MLCT lifetime of iron(II) complexes. We compare the [C2N4] donor set in the novel more octahedral carbene iron(II) complex [Fe(dpmi)2]2+12+ (Scheme 1) featuring facially coordinating dpmi ligands (di(pyridine-2-yl)(3-methylimidazol-2-yl)methane) that form six-membered chelate rings with distorted [Fe(CNC)2]2+ and [Fe(CNN)2]2+ complexes possessing five-membered chelate rings in terms of the resulting 3MLCT lifetime. The distorted [Fe(CNC)2]2+ and [Fe(CNN)2]2+ complexes with five-membered rings exhibit 3MLCT lifetimes below 0.1 ps.17 We demonstrate that the more octahedral and rigid complex geometry increases the lifetime even beyond that of a [C4N2] donor set in distorted carbene pyridine iron(II) complexes (τ = 9, 8.1, <0.3 ps for [Fe(CNC)2]2+ with Me, iPr, tBu N-substituents).16,17 Consequently, the higher octahedricity and rigidity of 12+ has a similar boosting effect as two strong carbene σ-donors in distorted [Fe(CNC)2]2+ complexes.
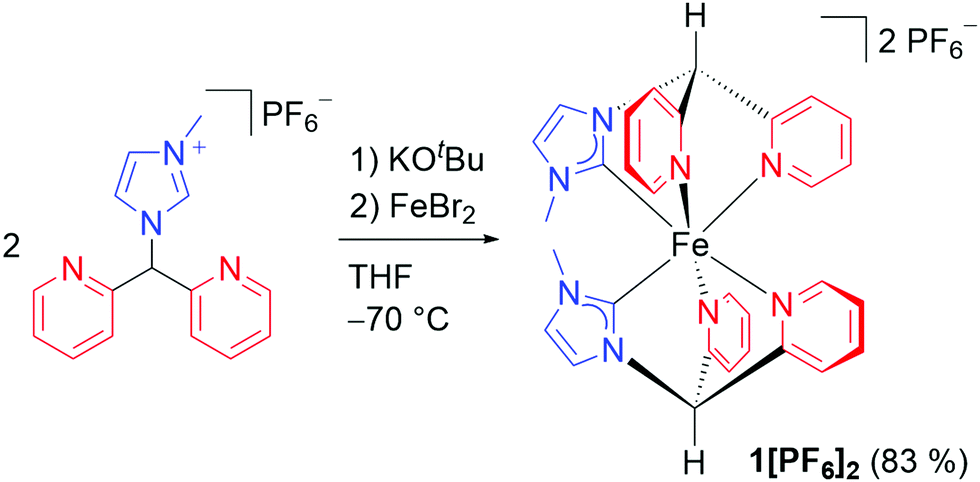 | ||
| Scheme 1 Synthesis of 1[PF6]2 starting from the pro-ligand [Py2MeImH][PF6].32 | ||
The tridentate facially coordinating CNN ligand derived from [Py2MeImH][PF6]32 after deprotonation forms six-membered chelate rings with iron(II) in the homoleptic complex cation 12+ (Scheme 1). Complex 12+ with a [C2N4] donor set was characterised by IR, NMR spectroscopy, ESI+ mass spectrometry and elemental analysis (ESI). The 12 and 15 1H and 13C NMR resonances observed (ESI) are consistent with a cis configuration of the carbene donors and the expected destabilising trans influence in a conceivable trans configuration. Six Fe–C/N atom distances of 1.99(8) Å were determined by iron K-edge EXAFS of 1[PF6]2 (ESI). Four Fe⋯N distances of 2.79(4) Å fit to the four NHC nitrogen atoms. The averaged Fe–C/N distances obtained by the EXAFS experiment agree with a structural model derived from quantum chemical calculations of low-spin 12+ (DFT CPCM(acetonitrile)-RIJCOSX-B3LYP-D3BJ-ZORA/def2-TZVP; averaged Fe–C/N distance of 1.999 Å and averaged Fe⋯Ncarbene distances of 2.943 Å; ESI). The pre-edge peak at 7113.3 eV in the iron K-edge X-ray absorption spectrum33 and the NMR data of 1[PF6]2 confirm the low-spin state of the iron(II) centre. The energies of the pre-peak and the near-edge shoulders around 7120 and 7123 eV of 12+ (ESI) are essentially identical to that of a [Fe(CNN)2]2+ complex with cis-positioned carbene donors in a more distorted geometry (7113.3, 7120, 7123 eV).17
The electronic absorption spectrum of 12+ in acetonitrile shows two absorption bands at 415 nm (ε = 17310 L mol−1 cm−1) and 500 nm (ε = 19390 L mol−1 cm−1) (Fig. 1a). Their intensities agree with allowed charge transfer transitions and exceed those of other carbene and pyridine iron(II) complexes, e.g. 700 nm (2000 L mol−1 cm−1), 508/557 nm (4700/7000 L mol−1 cm−1), 503/538 nm (9800/8600 L mol−1 cm−1) and 551 nm (7000 L mol−1 cm−1) for [Fe(btz)3]2+,20 [Fe(CNN)2]2+, [Fe(CNC)(tpy)]2+ and [Fe(tpy)2]2+, respectively.17 According to time-dependent DFT (TDDFT) calculation and charge transfer number analysis,34 these two intense bands consist of dFe → πpyridine1MLCT transitions, while dFe → πcarbene1MLCT transitions appear at higher energy (ESI). Less allowed transitions are calculated at lower energy. The charge transfer number analysis assigns mainly 1MC character to these bands, suggesting that 1MC states are lower in energy than 1MLCT states.
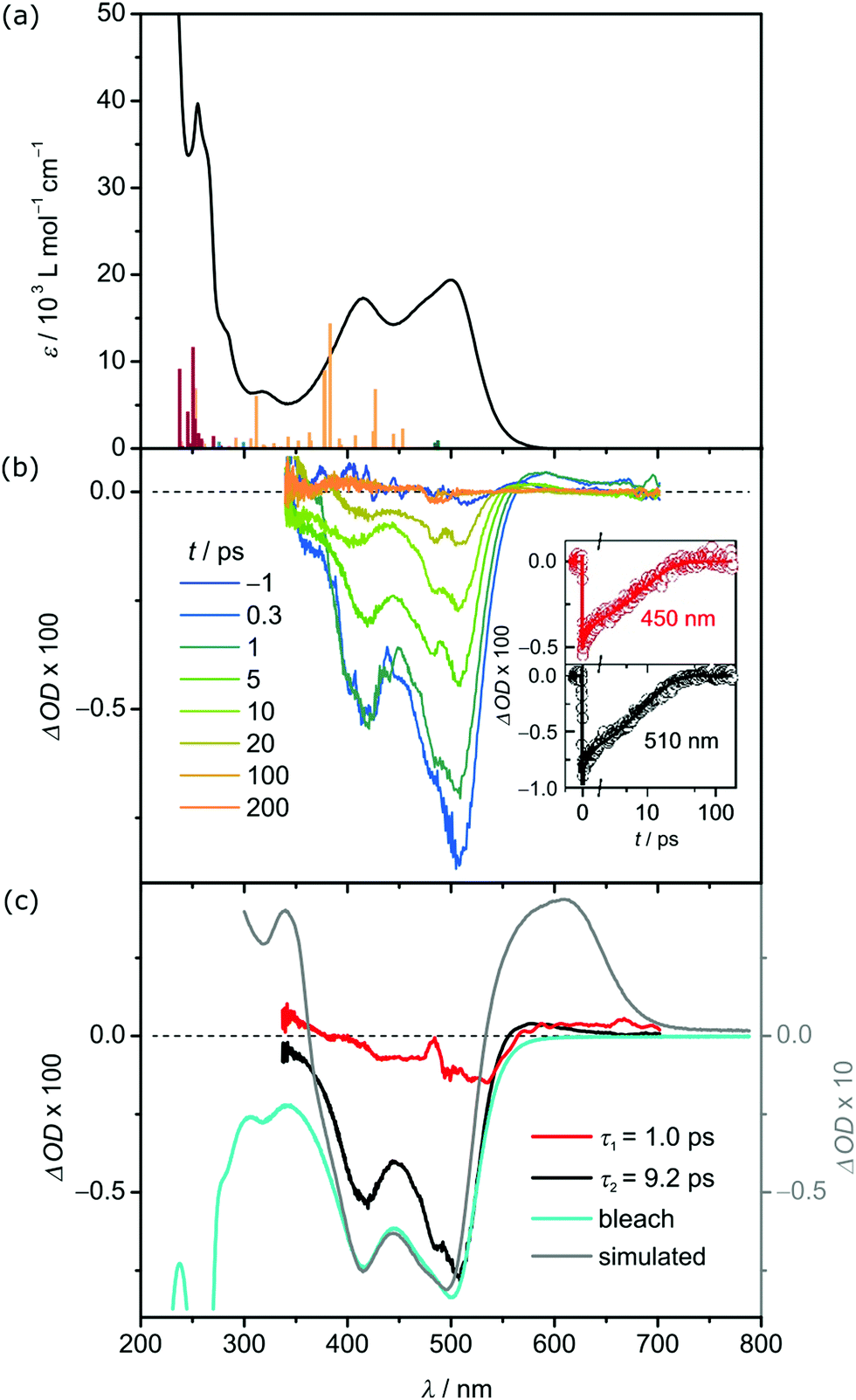 | ||
| Fig. 1 (a) UV/Vis absorption spectrum of 1[PF6]2 (black) in deaerated acetonitrile and TDDFT calculated transitions of 12+ with the colour code indicating the character of the transition according to charge transfer analysis (dark green: MC, orange: MLCT, dark red: LL’CT). (b) Transient absorption spectra of 1[PF6]2 in deaerated acetonitrile at selected time points from 0.3 to 200 ps after excitation at 490 nm. The insets show time traces probed at 450 nm (red) and 510 nm (black) and corresponding fits. (c) Decay associated amplitude spectra (DAS) of the 1.0 ps (red) and 9.2 ps (black) decay component compared to the scaled bleach (blue) and the difference spectrum35 between 13+ and 12+ (grey). | ||
The iron(II) complex 12+ is reversibly oxidised to 13+ at E1/2 = 0.26 V vs. ferrocene in the cyclic voltammogram (MeCN, [nBu4N][PF6], ESI). Spectro-electrochemical oxidation of 12+ to 13+ bleaches the MLCT absorption bands while two new bands appear at 522 and 610 nm (ESI). These are ascribed to carbene/pyridine-to-iron charge transfer transitions (LMCT). Isosbestic points form at 253, 267, 356 and 544 nm confirming the reversible nature of the oxidation process. Chemical oxidation of orange 12+ to blue 13+ using [NO][PF6] as oxidant is successful as well as supported by an identical UV/Vis spectral pattern (ESI).
Transient absorption (TA) spectroscopy on 1[PF6]2 in deaerated MeCN discloses the excited state dynamics after excitation at 490 nm (MLCT). Fig. 1b shows the TA spectra recorded at selected time delays following excitation at 490 nm. The TA spectra display two negative bands at 420 and 505 nm, which correspond to the ground state bleach (GSB), and a weaker excited state absorption (ESA) at approximately 560 nm. All signals decay on the 10 ps time scale (Fig. 1b, insets).
The dynamics is analysed by applying a global, triple-exponential fit, yielding a dominant decay component with a time constant of τ2 = 9.2 ps and a weaker one of τ1 = 1.0 ps. The third contribution exhibits a lifetime of only a few femtoseconds, which is much shorter than the actual time resolution, and resembles an artefact at time zero due to cross phase modulation. Fig. 1c compares the decay associated amplitude spectra (DAS) of the two relevant components τ1 and τ2 to the scaled bleach and the difference spectrum between 13+ and 12+. The DAS of the dominant τ2 = 9.2 ps component is quite similar to the bleach but exhibits additional ESA contributions in the wavelength range 550–650 nm. The difference spectrum indicates that oxidation of iron(II) to iron(III) causes such an additional absorption. Below 400 nm the TA signal approaches zero with decreasing wavelength in contrast to the bleach pointing also there to an additional ESA contribution. This too, would be in line with an oxidation of the iron center. Accordingly, the 9.2 ps component describes the decay of a state involving iron(III) suggesting that it is the MLCT state.30 The absorption band of 13+ at 610 nm and consequently the ESA of 12+ are of ligand→iron(III) LMCT character. The DAS of the 1.0 ps component is weak and exhibits a shape, which has some similarities with the dominant contribution but is slightly red shifted (see Fig. 1c). Therefore, it is assigned to energy redistribution processes, e.g. charge localisation in the ligand, which cause a blue shift of the TA spectra.
With this interpretation and literature precedent,15–17,35 the DAS within the first few picoseconds are assigned to the 3MLCT state of 12+. Intersystem crossing from 1MLCT to 3MLCT is faster than the time resolution of our instrument (<100 fs). Based on the observed decay of the TA spectra, relaxation of the 3MLCT state back to the ground state occurs either directly or via an intermediate state that is only transiently populated and thus not observed. Typically, 3MC and 5MC states serve as deactivating pathways for iron(II) complexes.5–8 Iron(II) complexes with the 5MC state as lowest excited state possess high 5MC lifetimes up to several nanoseconds, for example the 5T2–1A1 ground state recovery time of [Fe(tpy)2]2+ is 4 ns.36,37 On the other hand, complexes with the 3MC state lower than the 5MC state possess 3MC lifetimes of only a few ps and sometimes even less. For example, [Fe(CNC)2]2+ complexes with methyl and isopropyl N-substituents show 3MC lifetimes <2 ps and <8 ps, significantly shorter than the 3MLCT lifetime.16,17 A similar situation might be operative in 12+ with only two carbene ligands: the 5MC state is higher in energy than the 3MC state which decays rapidly to the ground state and is consequently not observed, similar to [Fe(CNC)2]2+.19
To substantiate this interpretation, the lowest triplet and quintet MC states of 12+ were calculated by DFT (Fig. 2). The 3MC state is slightly lower in energy than the 5MC state. In the triplet state, the two Fe–N distances of the trans positioned pyridines are strongly elongated from 2.013/2.015 Å to 2.348/2.358 Å, while the other Fe–C/N distances are hardly affected. This is consistent with the population of the dz2 orbital with the z-axis experiencing the weakest ligand field strength. In the 5MC high spin-state, all Fe–C/N distances are elongated due to the population of the dz2 and dx2−y2 orbitals. The large distortions impose a reorganisation barrier between the nearly degenerate 3MC and 5MC states. This substantiates the interpretation that the 5MC state is by-passed and that the 3MC state undergoes ISC to the ground state. Attempts to optimise the 3MLCT state by DFT without constraints were unsuccessful as all optimisation attempts converged to the 3MC state. By constraining all Fe–C/N bond distances to 1.9 Å, convergence to a 3MLCT state with spin density at the iron and a pyridine ring and an energy of 2.33 eV (532 nm) was achieved. This estimated 3MLCT energy is consistent with the experimental 1MLCT state energy of 2.48 eV (500 nm). Assuming that all MLCT states are rather nested with the ground state,19 population transfer from the 1/3MLCT states to the 3MC state is burdened with a reorganisation energy. In combination with the comparably high 3MC energy, this barrier accounts for the high 3MLCT lifetime of 12+ of 9.2 ps. Such a high lifetime of a 3MLCT state in iron(II) complexes has been only realised so far using four carbene donors instead of only two in 12+.16,17
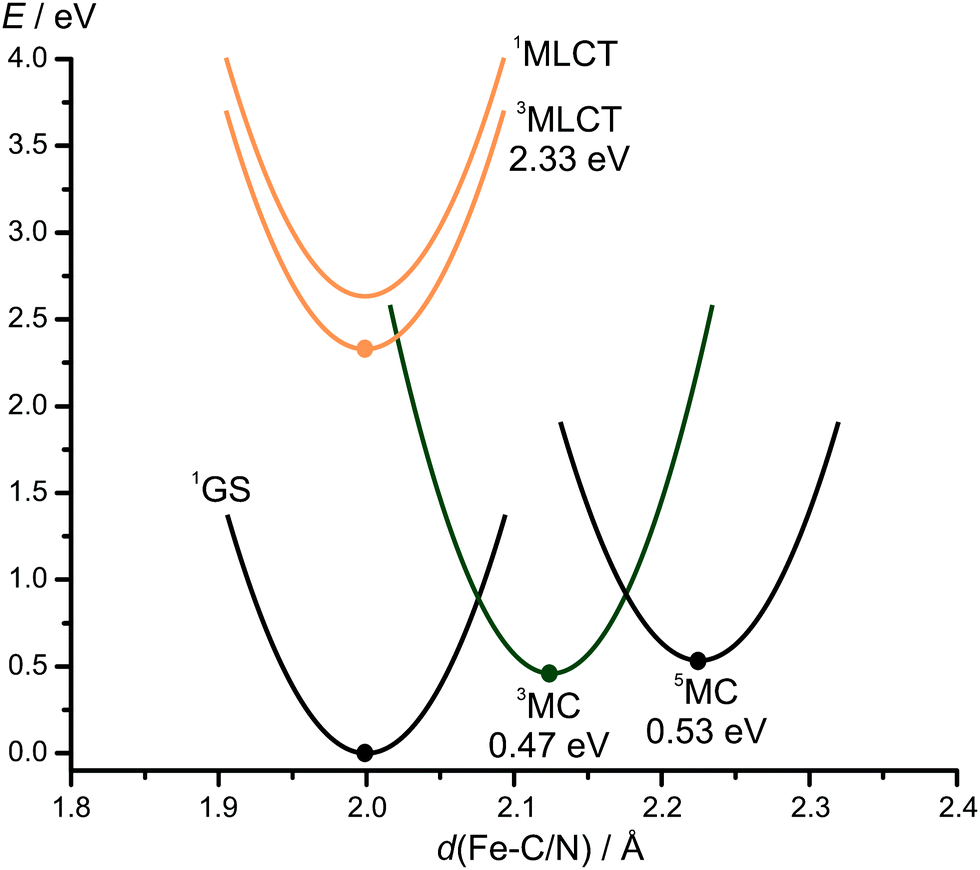 | ||
| Fig. 2 Summary of the quantum chemically calculated energetics of 12+ as obtained from DFT calculations (•) or experimental data (1MLCT), with parabolic energy surfaces sketched qualitatively as visual guides for discussion purposes. | ||
Emission from the 3MLCT state after excitation of 12+ at 413 or 500 nm at room temperature in MeCN or at 77 K in a frozen nPrCN glass was not observed indicating that the 3MLCT–3MC barrier can efficiently be overcome even at 77 K.
Carbenes also stabilise low-spin iron(III) and 2LMCT states of iron(III) complexes can be photoactive.30,38 Consequently, emission of the iron(III) complex 13+, prepared in situ by oxidation of 12+ with [NO][PF6], was probed by exciting at 524 and 609 nm at room temperature and at 77 K. However, no 2LMCT fluorescence of 13+ was detected with our instrument.
The iron(II) complex [Fe(dpmi)2]2+12+ with a two carbene/four pyridine [C2N4] donor set and a high octahedral symmetry enabled by six-membered chelate rings in a rigid environment exhibits a high 3MLCT lifetime of 9.2 ps. This value compares to lifetimes of iron(II) complexes with a four carbene/two pyridine [C4N2] donor set in a more distorted environment with five-membered chelate rings. This observation validates the high symmetry concept to increase the energy of MC states by a better metal–ligand orbital overlap. Deactivation of the 3MLCT state of 12+ likely occurs via the tetragonally distorted 3MC state by-passing the 5MC state resembling the photodynamics of classical polypyridine ruthenium(II) complexes.19 The combination of six-membered chelates and strong σ-donating carbenes paves the way to photoactive, luminescent and solar energy-converting iron(II) complexes with long 3MLCT lifetimes. A future challenge will be the increasingly facile FeII/FeIII oxidation with a high number of carbene donors and the smaller MLCT extinction coefficient in all-carbene complexes lacking π-accepting ligands.20,30,37
TR performed the synthesis, ground state characterisation and the DFT calculations. AK and SL measured and interpreted the TA data. RS and MB measured and interpreted the XAS data. KH designed and supervised the project, analysed the data and wrote the manuscript.
Financial support from the Deutsche Forschungsgemeinschaft [Priority Program SPP 2102 “Light-controlled reactivity of metal complexes” (HE 2778/14-1, BA 4467/7-1, LO 714/11-1)] is gratefully acknowledged. Parts of this research were conducted using the supercomputers MOGON and Elwetritsch and advisory services offered by Johannes Gutenberg university Mainz (http://www.hpc.uni-mainz.de) and TU Kaiserslautern (https://elwe.rhrk.uni-kl.de), which are members of the AHRP. PETRA III is kindly acknowledged for provision of beamtime at beamline P65.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057 RSC.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140(42), 13522 CrossRef CAS PubMed.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49 CrossRef CAS.
- B. M. Hockin, C. Li, N. Robertson and E. Zysman-Colman, Catal. Sci. Technol., 2019, 9, 889 RSC.
- O. S. Wenger, Chem. – Eur. J., 2019, 25, 6043 CrossRef CAS PubMed.
- Y. Liu, P. Persson, V. Sundström and K. Wärnmark, Acc. Chem. Res., 2016, 49, 1477 CrossRef CAS PubMed.
- S. Kaufhold and K. Wärnmark, Catalysts, 2020, 10, 132 CrossRef CAS.
- B. C. Paulus, S. L. Adelman, L. L. Jamula and J. K. McCusker, Nature, 2020, 582, 214 CrossRef CAS PubMed.
- J. K. McCusker, Science, 2019, 363, 484 CrossRef CAS PubMed.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Photochemistry and Photophysics of Coordination Compounds: ruthenium, Topics in Current Chemistry, Springer, Berlin, Heidelberg, 2007, vol. 280, p. 117 Search PubMed.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Photochemistry and Photophysics of Coordination Compounds: iridium, Topics in Current Chemistry, Springer, Berlin, Heidelberg, 2007, vol. 281, p. 143 Search PubMed.
- L. L. Jamula, A. M. Brown, D. Guo and J. K. McCusker, Inorg. Chem., 2014, 53, 15 CrossRef CAS PubMed.
- A. K. C. Mengel, C. Förster, A. Breivogel, K. Mack, J. R. Ochsmann, F. Laquai, V. Ksenofontov and K. Heinze, Chem. – Eur. J., 2015, 21, 704 CrossRef CAS PubMed.
- A. K. C. Mengel, C. Bissinger, M. Dorn, O. Back, C. Förster and K. Heinze, Chem. – Eur. J., 2017, 23, 7920 CrossRef CAS PubMed.
- M. Darari, E. Domenichini, A. Francés-Monerris, C. Cebrián, K. Magra, M. Beley, M. Pastore, A. Monari, X. Assfeld, S. Haacke and P. C. Gros, Dalton Trans., 2019, 48, 10915 RSC.
- Y. Liu, T. C. B. Harlang, S. E. Canton, P. Chábera, K. Suárez-Alcántara, A. Fleckhaus, D. A. Vithanage, E. Göransson, A. Corani, R. Lomoth, V. Sundström and K. Wärnmark, Chem. Commun., 2013, 49, 6412 RSC.
- P. Zimmer, L. Burkhardt, A. Friedrich, J. Steube, A. Neuba, R. Schepper, P. Müller, U. Flörke, M. Huber, S. Lochbrunner and M. Bauer, Inorg. Chem., 2018, 57, 360 CrossRef CAS PubMed.
- T. Harlang, Y. Liu, O. Gordivska, L. Fredin, C. Ponseca Jr., P. Huang, P. Chábera, K. Kjær, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Perrson, V. Sundström and K. Wärnmark, Nat. Chem., 2015, 7, 883 CrossRef CAS PubMed.
- L. A. Fredin, M. Pápai, E. Rozsályi, G. Vanko, K. Wärnmark, V. Sundström and P. Persson, J. Phys. Chem. Lett., 2014, 5, 2066 CrossRef CAS PubMed.
- P. Chábera, K. Kjær, O. Prakash, A. Honarfar, Y. Liu, L. Fredin, T. Harlang, S. Lidin, J. Uhlig, V. Sundström, R. Lomoth, P. Perrson and K. Wärnmark, J. Phys. Chem. Lett., 2018, 9, 459 CrossRef PubMed.
- M. Abrahamsson, M. Jäger, T. Österman, L. Eriksson, P. Persson, H.-C. Becker, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2006, 128, 12616 CrossRef CAS PubMed.
- M. Abrahamsson, H.-C. Becker, L. Hammarström, C. Bonnefous, C. Chamchoumis and R. P. Thummel, Inorg. Chem., 2007, 46, 10354 CrossRef CAS PubMed.
- M. Abrahamsson, M. Jäger, R. J. Kumar, T. Österman, P. Persson, H.-C. Becker, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2008, 130, 15533 CrossRef CAS PubMed.
- A. Breivogel, C. Förster and K. Heinze, Inorg. Chem., 2010, 49, 7052 CrossRef CAS PubMed.
- A. Breivogel, M. Meister, C. Förster, F. Laquai and K. Heinze, Chem. – Eur. J., 2013, 19, 13745 CrossRef CAS PubMed.
- M. Dorn, J. Kalmbach, P. Boden, A. Päpcke, S. Gómez, C. Förster, F. Kuczelinis, L. M. Carrella, L. Büldt, N. Bings, E. Rentschler, S. Lochbrunner, L. González, M. Gerhards, M. Seitz and K. Heinze, J. Am. Chem. Soc., 2020, 142, 7947 CrossRef CAS PubMed.
- S. Treiling, C. Wang, C. Förster, F. Reichenauer, J. Kalmbach, P. Boden, J. P. Harris, L. Carrella, E. Rentschler, U. Resch-Genger, C. Reber, M. Seitz, M. Gerhards and K. Heinze, Angew. Chem., Int. Ed., 2019, 58, 18075 CrossRef CAS PubMed.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572 CrossRef CAS PubMed.
- J. P. Harris, C. Reber, H. E. Colmer, T. A. Jackson, A. P. Forshaw, J. M. Smith, R. A. Kinney and J. Telser, Can. J. Chem., 2020, 98, 250 CrossRef CAS.
- K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K.-E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Science, 2019, 363, 249 CrossRef PubMed.
- M. Darari, A. Francés-Monerris, B. Marekha, A. Doudouh, E. Wenger, A. Monari, S. Haacke and P. C. Gros, Molecules, 2020, 25, 5991 CrossRef CAS PubMed.
- V. Tran, K. E. Allen, M. Garcia Chavez, C. Aaron, J. J. Dumais, J. T. York and E. C. Brown, Polyhedron, 2018, 147, 131 CrossRef CAS.
- G. Vankó, T. Neisius, G. Molnár, F. Renz, S. Kárpáti, A. Shukla and F. M. F. de Groot, J. Phys. Chem. B, 2006, 110, 11647 CrossRef PubMed.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- A. M. Brown, C. E. McCusker and J. K. McCusker, Dalton Trans., 2014, 43, 17635 RSC.
- J. K. McCusker, A. L. Rheingold and D. N. Hendrickson, Inorg. Chem., 1996, 35, 2100 CrossRef CAS.
- A. Hauser, C. Enachescu, M. L. Daku, A. Vargas and N. Amstutz, Coord. Chem. Rev., 2006, 250, 1642 CrossRef CAS.
- P. Chábera, Y. Liu, O. Prakash, E. Thyrhaug, A. El Nahhas, A. Honarfar, S. Essén, L. A. Fredin, T. C. B. Harlang, K. S. Kjær, K. Handrup, F. Ericson, H. Tatsuno, K. Morgan, J. Schnadt, L. Häggström, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, Nature, 2017, 543, 695 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, synthesis, spectral data, computational data. See DOI: 10.1039/d1cc02173g |
| This journal is © The Royal Society of Chemistry 2021 |