
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric autocatalysis triggered by triglycine sulfate with switchable chirality by altering the direction of the applied electric field†
Tsuneomi
Kawasaki
 a,
Yoshiyasu
Kaimori
a,
Seiya
Shimada
a,
Natsuki
Hara
a,
Susumu
Sato
a,
Kenta
Suzuki
a,
Toru
Asahi
b,
Arimasa
Matsumoto‡
a and
Kenso
Soai
*ac
a,
Yoshiyasu
Kaimori
a,
Seiya
Shimada
a,
Natsuki
Hara
a,
Susumu
Sato
a,
Kenta
Suzuki
a,
Toru
Asahi
b,
Arimasa
Matsumoto‡
a and
Kenso
Soai
*ac
aDepartment of Applied Chemistry, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: soai@rs.kagu.tus.ac.jp
bDepartment of Life Science and Medical Bioscience, Waseda University (TWIns), Wakamatsu-cho, Shinjuku-ku, Tokyo 162-8480, Japan
cResearch Organization for Nano & Life Innovation, Waseda University, Wasedatsurumaki-cho, Shinjuku-ku, Tokyo, 162-0041, Japan
First published on 13th May 2021
Abstract
Triglycine sulfate (TGS) acts as a chiral trigger for asymmetric autocatalysis with amplification of enantiomeric excess, i.e., the Soai reaction. Therefore, molecular chirality of highly enantioenriched organic compounds is controlled by a ferroelectric crystal TGS, whose polarization is altered by an electric field.
Control of molecular chirality by the application of physical force is one of the most challenging topics in science,1–3 and this approach can provide a new methodology for enantioselective synthesis.4–7 The induction of chirality by physical force8 is an important topic to consider with respect to the origin of biological homochirality,9 which continues to attract tremendous attention from scientists in many fields of research.10–12 An electromagnetic force is often considered a possible inducer of molecular chirality because electromagnetic waves can directly interact with organic molecules. Actually, chiral electromagnetic force, circularly polarized light (CPL), can induce enantioselective decomposition13 and reactions14 of organic compounds, and asymmetric amplification during the crystallization15 or asymmetric autocatalysis16,17 can provide chiral organic products with a high enantiomeric excess (ee) initiated by CPL irradiation. Furthermore, recent observations have experimentally demonstrated that the combination of path direction of linearly polarized light and magnetic field can provide chiral effects leading to the formation of enantioenriched compounds.3,18 To our knowledge, there has been no apparent example of the control of molecular chirality in enantioselective reaction by a ferroelectric crystal, whose chirality is induced by the application of a static electric field, an achiral physical force.
Here, we describe the asymmetric autocatalysis of 5-pyrimidyl alkanol (the Soai reaction)12,16,19–24 initiated with ferroelectric triglycine sulfate (TGS: (NH2CH2COOH)3·H2SO4) crystal, whose handedness is controlled by the application of a static electric field (Fig. 1).
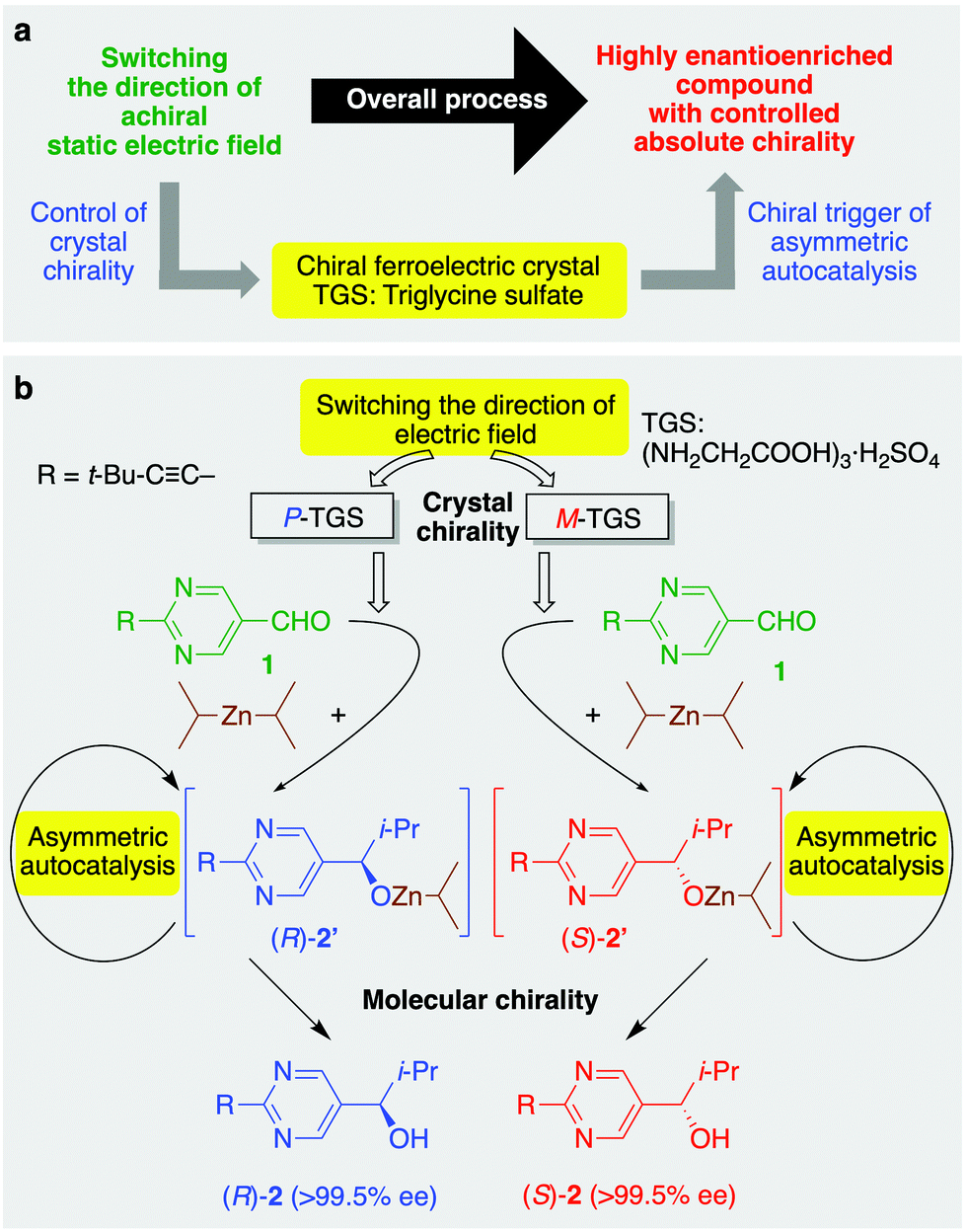 | ||
| Fig. 1 Concept and scheme of the present work: (a) control of the absolute molecular chirality by selecting the direction of the static electric field applied to triglycine sulfate (TGS) and asymmetric autocatalysis with amplification of ee. (b) Asymmetric autocatalysis of 5-pyrimidyl alkanol 2 induced by P- and M-TGS as a heterogeneous chiral trigger, leading to highly enantioenriched (R)- and (S)-alkanol 2, respectively. | ||
We first reported asymmetric autocatalysis with amplification of ee as a real chemical reaction.16 When diisopropylzinc (i-Pr2Zn) addition to pyrimidine-5-carbaldehyde 1 was performed in the presence of a catalytic amount of pyrimidyl alkanol 2 with a very low ee (ca. 0.00005% ee), asymmetric autocatalysis could enhance the ee significantly to produce a final product with very high ee (>99.5% ee).25 Asymmetric autocatalysis, named as the Soai reaction,19,20 has enormous power to recognize and amplify small chiral imbalances such as carbon isotope chirality7 and crystal chirality;26,27 therefore, asymmetric autocatalysis can connect the proposed origins of chirality with enantiomerically enriched organic compounds.12 To induce molecular chirality using the controlled chirality of TGS, asymmetric autocatalysis of 5-pyrimidyl alkanol 2 (its isopropylzinc alkoxide 2′) in the addition reaction of i-Pr2Zn to pyrimidine-5-carbaldehyde 1 was performed with TGS operating as a chiral trigger (Fig. 1b). We assumed that this reaction would allow the crystal chirality of TGS to be discriminated and afford highly enantioenriched product with the handedness corresponding to TGS.
TGS (Fig. 2a) is a well-known ferroelectric crystal.28 Crystals of this compound exhibit spontaneous electric polarization and polarities, which can be controlled by the application of an external static electric field. Moreover, crystals of TGS have chirality even though the components including glycine and sulfate ions are achiral. However, chiral crystals of TGS have rarely been used as a chiral source for enantioselective reactions despite their wide application as a ferroelectric material.
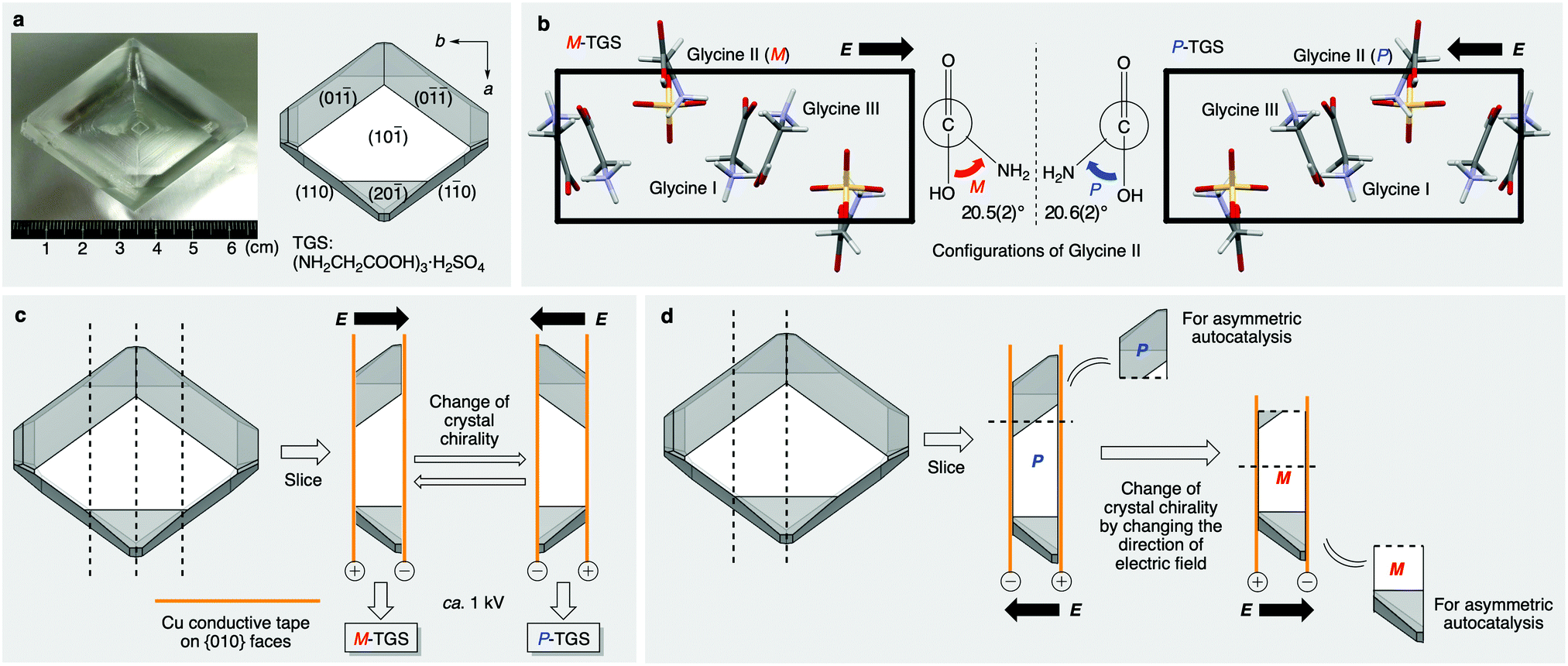 | ||
| Fig. 2 The chirality of TGS and its control by application of a static electric field. (a) Morphology of a single crystal of TGS [(NH2CH2COOH)3·H2SO4]. (b) Comparison of the enantiomorphs of TGS at the molecular level and the twisted chiral structure of one glycine (glycine II) among three glycines in the crystal lattice. (c) The absolute correlation between the direction of the applied electric field and the crystal chirality of the resulting TGS. (d) The control of the chirality of TGS by switching the direction of the applied static electric field to one specific piece of TGS. | ||
TGS crystal belongs to the monoclinic P21 space group (a = 9.44 Å, b = 12.6 Å, c = 5.74 Å, β = 110°),29 which does not have inversion or mirror symmetry (Fig. 2b and Fig. S1, S2, ESI†). There are three symmetrically independent glycine molecules in a unit cell. One glycine molecule (glycine II) exists in the zwitterion form and has a large torsion angle of ca. +21° (P-conformation) or −21° (M-conformation) for the N–C–C–O bond, whereas the other two glycine molecules (glycines I and III) exist as almost planar structures in the protonated form. The twisted glycine II induces crystal polarity because no symmetry operator can invert or rotate the b-axis direction in the P21 space group. Typically, TGS crystals that are grown from an aqueous solution are twinned crystals of both domains, in which spontaneous polarizations align parallelly or anti-parallelly to the b-axis. The ferroelectricity is induced by expanding and shrinking these twin domain areas in the crystal in an electric field. The important point is that the symmetrical relationship between these two domains is not a 180° rotation but a reflection.30 This means that the electric field application also inverts the crystal chirality in addition to the polarity under an electric field.
To examine the control of chirality of TGS, we prepared single crystals of TGS and undertook an absolute structure determination by single-crystal X-ray diffraction analysis after the application of an electric field. The naturally grown TGS obtained from the slow evaporation of an aqueous solution of sulfuric acid and glycine was elongated in the [010] direction and had well-developed {10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) } faces with characteristic {0}, {110} and {20} faces (Fig. 2a). When the crystal is placed with the (01) and (0) faces up and triangle (20) face down against the large front (10) surface, the polar b-axis was directed and left in this axis setting. The crystal was sliced perpendicular to the b-axis to give {010} surfaces (Fig. 2c). And a static electric field was applied to this by attaching conductive Cu tape on the {010} faces for poling the crystal. The chirality of the resulting crystal was confirmed by anomalous dispersion measurements in single-crystal X-ray diffraction analysis for several points of the crystal, which revealed that the absolute crystal chirality of TGS was well-controlled by this method even in the bulk size slice of the crystal (ca. 4 cm long and 5–7 mm thick). When the electric field was applied antiparallel to the b-axis, the handedness of the resulting crystal was the M-conformation, and generation of the opposite P-conformation was observed when the electric field was applied parallel to the b-axis. The Flack parameter with inversion twin refinement is typically estimated to be around 0.1–0.2 with the standard uncertainty around 0.1. The results mean that the glycine molecules predominantly twist in one direction and have controlled chirality by the electric field (ca. 60–80% crystal ee).
} faces with characteristic {0}, {110} and {20} faces (Fig. 2a). When the crystal is placed with the (01) and (0) faces up and triangle (20) face down against the large front (10) surface, the polar b-axis was directed and left in this axis setting. The crystal was sliced perpendicular to the b-axis to give {010} surfaces (Fig. 2c). And a static electric field was applied to this by attaching conductive Cu tape on the {010} faces for poling the crystal. The chirality of the resulting crystal was confirmed by anomalous dispersion measurements in single-crystal X-ray diffraction analysis for several points of the crystal, which revealed that the absolute crystal chirality of TGS was well-controlled by this method even in the bulk size slice of the crystal (ca. 4 cm long and 5–7 mm thick). When the electric field was applied antiparallel to the b-axis, the handedness of the resulting crystal was the M-conformation, and generation of the opposite P-conformation was observed when the electric field was applied parallel to the b-axis. The Flack parameter with inversion twin refinement is typically estimated to be around 0.1–0.2 with the standard uncertainty around 0.1. The results mean that the glycine molecules predominantly twist in one direction and have controlled chirality by the electric field (ca. 60–80% crystal ee).
A mixture of pyrimidine-5-carbaldehyde 131,32 and TGS was ground into a powder using a pestle and mortar, and then i-Pr2Zn was added dropwise. Furthermore, asymmetric autocatalysis by subsequent addition of aldehyde 1 and i-Pr2Zn to the mixture gave highly enantioenriched 5-pyrimidyl alkanol 2 by the amplification of ee (Fig. 1b). When the reaction was performed in the presence of M-TGS, obtained from the application of an electric field parallel to the b-axis, (S)-pyrimidyl alkanol 2 was obtained with 65% ee (Table 1, series A, run 1). In contrast, P-TGS was produced as a result of the application of a static electric field from the direction antiparallel to the b-axis, giving oppositely configured (R)-alkanol 2 with 68% ee after the amplification of the ee by asymmetric autocatalysis (run 2). These results demonstrate that the direction of the static electric field can be used to control the molecular chirality of organic compounds through chiral ferroelectric TGS and asymmetric autocatalysis. It should be noted that P- and M-TGS used in the experiments detailed in runs 1 and 2 were generated from the one specific mother single crystal (crystal #1) by the application of oppositely directed static electric fields. The sense of enantioselectivity between TGS and alkanol 2 was reproducible, as shown in runs 3–6. Moreover, the ee of product 2 could be enhanced to near enantiopure levels (>99.5% ee) by conducting additional rounds of asymmetric autocatalysis, as shown in runs 7 and 8.
| Run | TGS | Pyrimidyl alkanol 2 | ||||
|---|---|---|---|---|---|---|
| Single-crystal batch | Direction of E | Chirality | Yield (%) | ee (%) | Config. | |
| a Additional rounds of asymmetric autocatalyses were performed to amplify the ee value significantly. b TGS crystal #5 was first applied with the static electric field parallel to the b-axis of TGS. The resulting P-TGS was used in run 9. The chirality of the same P-TGS crystal in run 9 was switched to M by applying the static electric field of the opposite direction of anti-parallel to the b-axis of TGS. c TGS crystal #6 was treated in the same manner with the footnote b. | ||||||
| Series A | ||||||
| 1 | #1 | → | M | 73 | 65 | S |
| 2 | #1 | ← | P | 62 | 68 | R |
| 3 | #2 | → | M | 41 | 43 | S |
| 4 | #2 | ← | P | 50 | 40 | R |
| 5 | #3 | → | M | 69 | 76 | S |
| 6 | #3 | ← | P | 74 | 39 | R |
| 7 | #4 | → | M | 67 (87)a | 44 (>99.5)a | S |
| 8 | #4 | ← | P | 80 (95)a | 54 (>99.5)a | R |
| Series B | ||||||
| 9 | #5 | ← | P | 91 | 92 | R |
| 10b | #5 | ← then → | M | 91 | 91 | S |
| 11 | #6 | ← | P | 85 | 88 | R |
| 12c | #6 | ← then → | M | 89 | 93 | S |
It was also found that the chirality of the same piece of TGS crystal is controlled repeatedly by applying the parallel and anti-parallel electric field successively (Fig. 2d and Table 1, series B). Thus, to confirm the stereochemical relationship between the direction of the electric field and the molecular handedness of the product of the asymmetric autocatalysis, switching experiments with chiral TGS and its use as a chiral trigger for asymmetric autocatalysis were conducted (Table 1, runs 9–12). After the application of an electric field parallel to the b-axis, half of the resulting P-crystal piece was used for the asymmetric autocatalysis affording (R)-product 2 with 92% ee (series B, run 9). The other piece was exposed to the electric field directed in the opposite direction, leading to its reconstruction as the opposite M-enantiomorph, which was also used as a chiral trigger for asymmetric autocatalysis. As a result, the enantioselectivity of the reaction was reversed to form the opposite (S)-alkanol 2 with a high 91% ee after significant amplification of the ee by asymmetric autocatalysis (run 10). The results were reproducible, as shown in runs 11 and 12. Therefore, the switch of the direction of the electric field is responsible for the change in the absolute configuration of the resulting product of asymmetric autocatalysis via ferroelectric TGS.
In summary, we have demonstrated that ferroelectric TGS, whose chirality is switchable by changing the direction of the applied static electric field, can induce a highly enantioselective synthesis through the significant amplification of ee by asymmetric autocatalysis of 5-pyrimidyl alkanol. The direction of an achiral static electric field and highly enantioenriched organic products were successfully linked with each other via the ferroelectric crystal of TGS; therefore, the present results provide new insight into the origin and amplification of biological homochirality.
The authors thank Yuko Araki, Kunihiko Hatase and Koichi Abe for the preliminary experiments. This work has been financially supported by Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (JSPS KAKENHI Grant Numbers 23685012, 26810026, 15H03781, 18H04518, 19K05482 and 19K22190, Grant-in-Aid for JSPS Research Fellow to Y. K. Number 15J03968) and by MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2012–2016.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418–3438 CrossRef PubMed.
- K.-H. Ernst, Phys. Status Solidi, 2012, 249, 2057–2088 CrossRef CAS.
- G. L. Rikken and E. Raupach, Nature, 2000, 405, 932–935 CrossRef CAS PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS.
- J. D. Horvath and A. J. Gellman, J. Am. Chem. Soc., 2001, 123, 7953–7954 CrossRef CAS PubMed.
- J. M. Ribo, J. Crusats, F. Sagués, J. Claret and R. Rubires, Science, 2001, 292, 2063–2066 CrossRef CAS PubMed.
- T. Kawasaki, Y. Matsumura, T. Tsutsumi, K. Suzuki, M. Ito and K. Soai, Science, 2009, 324, 492–495 CrossRef CAS PubMed.
- M. Avalos, R. Babiano, P. Cintas, J. L. Jimenez, J. C. Palacios and L. D. Barron, Chem. Rev., 1998, 98, 2391–2404 CrossRef CAS PubMed.
- M. Bolli, R. Micura and A. Eschenmoser, Chem. Biol., 1997, 4, 309–320 CrossRef CAS.
- I. Weissbuch and M. Lahav, Chem. Rev., 2011, 111, 3236–3267 CrossRef CAS PubMed.
- (a) D. K. Kondepudi and K. Asakura, Acc. Chem. Res., 2001, 34, 946–954 CrossRef CAS PubMed; (b) C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef PubMed.
- K. Soai, T. Kawasaki and A. Matsumoto, Acc. Chem. Res., 2014, 47, 3643–3654 CrossRef CAS PubMed.
- H. Nishino, A. Kosaka, G. A. Hembury, F. Aoki, K. Miyauchi, H. Shitomi, H. Onuki and Y. Inoue, J. Am. Chem. Soc., 2002, 124, 11618–11627 CrossRef CAS PubMed.
- (a) H. Kagan, A. Moradpour, J. F. Nicoud, G. Balavoine and G. Tsoucaris, J. Am. Chem. Soc., 1971, 93, 2353–2354 CrossRef CAS; (b) W. J. Bernstein, M. Calvin and O. Buchardt, J. Am. Chem. Soc., 1972, 94, 494–498 CrossRef CAS.
- W. L. Noorduin, A. C. Bode, M. van der Meijden, H. Meekes, A. F. van Etteger, W. J. P. van Enckevort, P. C. M. Christianen, B. Kaptein, R. M. Kellogg, T. Rasing and E. Vlieg, Nat. Chem., 2009, 1, 729–732 CrossRef CAS PubMed.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767–768 CrossRef CAS.
- (a) T. Kawasaki, M. Sato, S. Ishiguro, T. Saito, Y. Morishita, I. Sato, H. Nishino, Y. Inoue and K. Soai, J. Am. Chem. Soc., 2005, 127, 3274–3275 CrossRef CAS PubMed; (b) I. Sato, R. Sugie, Y. Matsueda, Y. Furumura and K. Soai, Angew. Chem., Int. Ed., 2004, 43, 4490–4492 CrossRef CAS PubMed.
- Y. Kitagawa, H. Segawa and K. Ishii, Angew. Chem., Int. Ed., 2011, 50, 9133–9136 CrossRef CAS PubMed.
- (a) T. Gehring, M. Quaranta, B. Odell, D. G. Blackmond and J. M. Brown, Angew. Chem., Int. Ed., 2012, 51, 9539–9542 CrossRef CAS PubMed; (b) L. Schiaffino and G. Ercolani, Angew. Chem., Int. Ed., 2008, 47, 6832–6835 CrossRef CAS PubMed.
- K. Mislow, Collect. Czech. Chem. Commun., 2003, 68, 849–864 CrossRef CAS.
- K. Micskei, G. Rabai, E. Gal, L. Caglioti and G. Palyi, J. Phys. Chem. B, 2008, 112, 9196–9200 CrossRef CAS PubMed.
- (a) M. E. Noble-Teran, J.-M. Cruz, J.-C. Micheau and T. Buhse, ChemCatChem, 2018, 10, 642–648 CrossRef CAS; (b) M. Funes-Maldonado, B. Sieng and M. Amedjkouh, Org. Lett., 2016, 18, 2536–2539 CrossRef CAS PubMed; (c) I. D. Gridnev and A. K. Vorobiev, ACS Catal., 2012, 2, 2137–2149 CrossRef CAS; (d) É. Dóka and G. Lente, J. Am. Chem. Soc., 2011, 133, 17878–17881 CrossRef PubMed.
- (a) S. V. Athavale, A. Simon, K. N. Houk and S. E. Denmark, Nat. Chem., 2020, 12, 412–423 CrossRef CAS PubMed; (b) O. Trapp, S. Lamour, F. Maier, A. F. Siegle, K. Zawatzky and B. F. Straub, Chem. – Eur. J., 2020, 26, 15871–15880 CrossRef CAS PubMed.
- Reviews: (a) K. Soai, T. Kawasaki and A. Matsumoto, Symmetry, 2019, 11, 694 CrossRef CAS; (b) K. Soai, Proc. Jpn. Acad., Ser. B, 2019, 95, 89–109 CrossRef CAS PubMed; (c) D. G. Blackmond, Chem. Rev., 2020, 120, 4831–4847 CrossRef CAS PubMed; (d) T. Buhse, J. M. Cruz, M. E. Noble-Terán, D. Hochberg, J. M. Ribó, J. Crusats and J.-C. Micheau, Chem. Rev., 2021, 121, 2147–2229 CrossRef CAS PubMed.
- I. Sato, H. Urabe, S. Ishiguro, T. Shibata and K. Soai, Angew. Chem., Int. Ed., 2003, 42, 315–317 CrossRef CAS PubMed.
- K. Soai, S. Osanai, K. Kadowaki, S. Yonekubo, T. Shibata and I. Sato, J. Am. Chem. Soc., 1999, 121, 11235–11236 CrossRef CAS.
- (a) T. Kawasaki, K. Jo, H. Igarashi, I. Sato, M. Nagano, H. Koshima and K. Soai, Angew. Chem., Int. Ed., 2005, 44, 2774–2777 CrossRef CAS PubMed; (b) H. Shindo, Y. Shirota, K. Niki, T. Kawasaki, K. Suzuki, Y. Araki, A. Matsumoto and K. Soai, Angew. Chem., Int. Ed., 2013, 52, 9135–9138 CrossRef CAS PubMed.
- (a) B. T. Matthias, C. E. Miller and J. P. Remeika, Phys. Rev., 1956, 104, 849–850 CrossRef CAS; (b) A. Lurio and E. Stern, J. Appl. Phys., 1960, 31, 1125–1126 CrossRef CAS; (c) S. N. Drozhdin and O. M. Golitsyna, Phys. Solid State, 2012, 54, 905–910 CrossRef CAS; (d) J. M. Hudspeth, D. J. Goossens, T. R. Welberry and M. J. Gutmann, J. Mater. Sci., 2013, 48, 6605–6612 CrossRef CAS.
- (a) S. Hoshino, Y. Okaya and R. Pepinsky, Phys. Rev., 1959, 115, 323–330 CrossRef CAS; (b) E. A. Wood and A. N. Holden, Acta Crystallogr., 1957, 10, 145–146 CrossRef CAS.
- J. Kobayashi, K. Uchino, H. Matsuyama and K. Saito, J. Appl. Phys., 1991, 69, 409–413 CrossRef CAS.
- T. Shibata, S. Yonekubo and K. Soai, Angew. Chem., Int. Ed., 1999, 38, 659–661 CrossRef CAS PubMed.
- Y. Kaimori, Y. Hiyoshi, T. Kawasaki, A. Matsumoto and K. Soai, Chem. Commun., 2019, 55, 5223–5226 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, crystallographic data, ortep drawings of P/M-triglycine sulfate and photo of powdered TGS. CCDC 1993618 and 1993609. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc02162a |
| ‡ Present address: Department of Chemistry, Biology and Environmental Science, Nara Women's University, Kita-Uoya Nishi-machi, Nara, 630-8506, Japan. |
| This journal is © The Royal Society of Chemistry 2021 |