
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed stereoselective domino arylation–acylation: an entry to chiral tetrahydrofluorenone scaffolds†
Petter
Dunås
 a,
Andrew J.
Paterson
a,
Gabriele
Kociok-Köhn
b,
Martin
Rahm
a,
Simon E.
Lewis
*cd and
Nina
Kann
*a
a,
Andrew J.
Paterson
a,
Gabriele
Kociok-Köhn
b,
Martin
Rahm
a,
Simon E.
Lewis
*cd and
Nina
Kann
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-41296 Gothenburg, Sweden. E-mail: kann@chalmers.se
bMaterials and Chemical Characterization Facility, Convocation Avenue, University of Bath, Bath, BA2 7AY, UK
cCentre for Sustainable Circular Technologies, Convocation Avenue, University of Bath, Bath, BA2 7AY, UK. E-mail: S.E.Lewis@bath.ac.uk
dDepartment of Chemistry, Convocation Avenue, University of Bath, Bath, BA2 7AY, UK
First published on 2nd June 2021
Abstract
A palladium-catalyzed domino arylation-cyclization of biocatalytically derived cyclic 1,3-dienes is demonstrated. The reaction introduces a high degree of structural complexity in a single step, giving access to tricyclic tetrahydrofluorenones with full regio- and stereoselectivity. The transformation proceeds through a novel acylation-terminated Heck-type sequence, and quantum chemical calculations indicate that C–H activation is involved in the terminating acylation step.
Palladium-catalyzed domino reactions provide an efficient means of rapidly constructing bi-, tri- and tetracyclic molecules,1 and have been applied in the formation of various natural products2 and heterocycles.1b,3 The tricyclic hydrofluorenone motif is well suited to benefit from such a synthesis and is present in a number of known bioactive compounds, such as taiwaniaquinol,4 asterogynin B,5 and kinamycin F6 (Fig. 1).
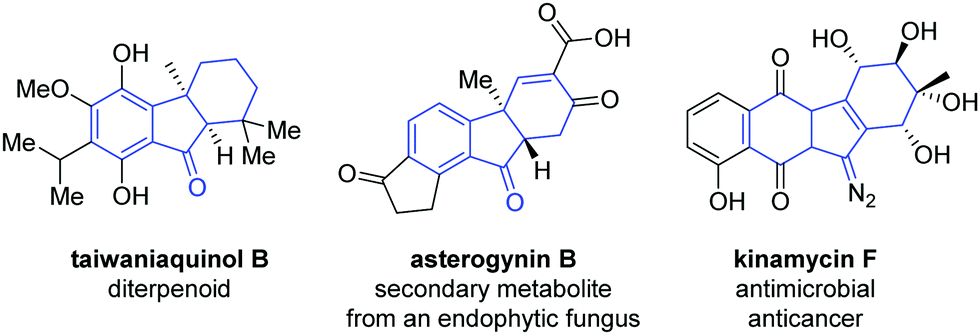 | ||
| Fig. 1 Hydrofluorenone-based natural produtcs. | ||
While palladium catalysis has been applied in this context,7 the use of domino reactions is as yet limited.8 Wu and Walsh have employed domino Heck/Tsuji–Trost reactions in the synthesis of chiral tetrahydrofluorenes,9 and asterogynin derivatives,10 but other reports are scarce.11 If the central ring of the tetrahydrofluorenone could be formed by a Pd-catalyzed intramolecular acylation, this could open up for new synthetic routes to such polycyclic structures. Acylation of an arylpalladium species has been described with aldehydes by Martin in the synthesis of benzocyclobutenones (Scheme 1a),12 while Solé,13 and Burke,14 cyclized heteroatom-tethered aryl halides with aldehydes. An sp3-palladium acylation, in the form of a domino nucleopalladation – acylation, has been reported by Liu and Lei (Scheme 1b).15 Heck-type arylation of alkynes and arynes with 2-halobenzaldehydes has been shown to trigger intramolecular acylation of the resultant alkenylpalladium species, as demonstrated by Heck, Larock and others (Scheme 1c).16 A few examples of nickel-catalyzed cyclizations between 2-halobenzaldehydes and alkenes or alkynes have also been reported.17 We here show that homochiral tetrahydrofluorenones can be formed in a single step using a Pd-catalyzed domino Heck-type carbopalladation–acylation reaction, starting from a (2-iodoaryl)-aldehyde and a chiral cyclohexadiene (Scheme 1d). The reaction presented herein is to our knowledge the only example of a cyclative Pd-catalyzed Heck-type domino alkene insertion-acylation.
 | ||
| Scheme 1 Pd-catalyzed intramolecular acylation to form fused cyclic compounds. | ||
Our investigation was triggered by the discovery that the Pd-catalyzed arylation of diene 1 with 2-iodobenzaldehyde result-ed in the formation of tricyclic tetrahydrofluorenone 2 (Scheme 2). Attempted use of other palladium or silver sources was not successful, while the amount of aryl halide could be lowered to 1.5 equiv with only a minor lowering of the yield (see ESI†).
 | ||
| Scheme 2 Formation of tricyclic tetrahydrofluorenone 2. | ||
Diene 1 derives from a biocatalytic arene oxidation (BAO) product, readily obtainable in multigram quantities via a dearomatizing oxidation of benzoic acid by R. eutropha B9 (Scheme 3).18 A range of other diene substrates were also prepared from the same BAO oxidation product by acetal protection of the diol followed by functional group conversion of the carboxylic acid.
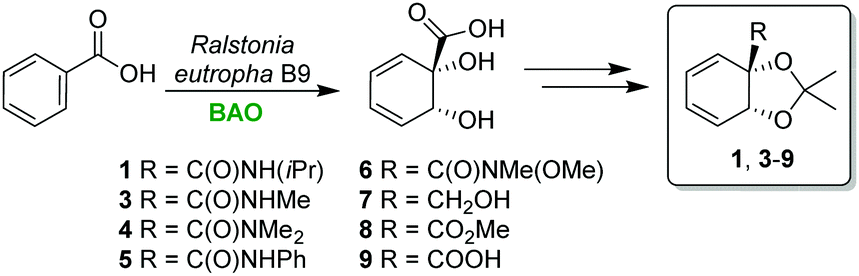 | ||
| Scheme 3 Biocatalytic arene oxidation (BAO) to access chiral dienes 1 and 3–9. | ||
Compounds 1 and 3–9 were then evaluated with 2-iodo-5-methoxybenzaldehyde as the aryl halide (Scheme 4). Methyl amide 3 performs well, affording 10 in 61% yield. Secondary amide 4 and aryl amide 5 are also compatible, producing 11 and 12 in somewhat lower yields. A better result (74%) is obtained with isopropylamide 1. Weinreb amide 14 provides a useful functionality for further transformations. Alcohol 15 could be formed by switching to DMF as the solvent, albeit in a low yield. For diene 8, with a pendant carboxylic acid, degradation of the diene starting material took place under the reaction conditions. For methyl ester 9, no conversion to product was seen, indicating that the amide hydrogen might have an activating effect in the reaction.
 | ||
| Scheme 4 aScope and limitations of diene and aryl halide. aIsolated yields. b100 °C in 1,4-dioxane. c100 °C in DMF. | ||
The scope of aryl halides was then evaluated with diene 1 under the same reaction conditions. With 2-iodobenzaldehyde, cyclization product 2 can be isolated in 61% yield. 2-Iodo-5-methoxybenzaldehyde affords 13 in 74% yield and the isomeric 2-iodo-4-methoxybenzaldehyde product 16 can be isolated in 58% yield using a higher reaction temperature. A higher temperature is also needed if the aryl iodide is substituted by a second halide or by a CF3-group (17–19), while dimethylated product 20 can be formed using the standard reaction conditions. Heteroaromatic aryl halides, containing a thiophene or indole moiety, proved unsuccessful. Likewise, the reaction seems limited to aryl iodides, with only trace amounts of 2 formed when 2-bromobenzaldehyde is used.
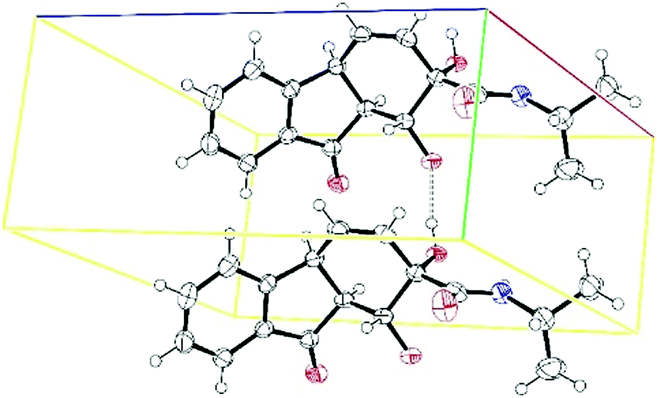
Acetonide deprotection to form 21 was also demonstrated, (Scheme 5). Diol 21 crystallizes readily, permitting structural elucidation by X-ray diffraction (Fig. 2).
 | ||
| Scheme 5 Acetonide deprotection of cyclization product 2. | ||
We here propose a mechanism in which, after the initial arylation of the diene through migratory insertion, the formed organopalladium intermediate undergoes an acylation by the adjacent aldehyde. As no non-cyclized Heck-product is detected, this acylation must be favoured over the β-hydride elimination forming 22 (Scheme 6, path C). In earlier work from our group, we have seen that β-hydride elimination is indeed favoured if no formyl group is present on the aryl halide.19 To form product 2, two pathways are possible: the cyclization could either result from an aldehyde C–H activation, followed by C–C bond formation via reductive elimination (path A). Alternatively, it might result from an aldehyde insertion into the Pd–C bond, followed by oxidation of the formed benzylic alkoxide by β-hydride elimination (path B).
 | ||
| Scheme 6 Potential mechanistic pathways. | ||
Both C–H activation12,20 and aldehyde insertion13,21 have been suggested in related aryl Pd-acylations. Rodrigo reported an aldehyde C–H activation by an alkylpalladium species leading to a decarbonylation,22 however, only one example of an acylation between a C(sp3)–Pd species and an aldehyde has been disclosed.15 An aldehyde insertion pathway was suggested in this case, but no mechanistic investigation was conducted to distinguish between these two pathways. As no distinction between the two mechanisms can be made based on our experiments, we have conducted a quantum chemical investigation using Density Functional Theory calculations. Calculations were performed at the SMD-B3LYP-D3/def2-TZVPD//def2-SVPD level of theory using Gaussian 16.23 Computational details, energies and optimized geometries of all structures can be found in the ESI.†
The formation of product 2 without added ligand was chosen as a model reaction to limit the cost of the calculations. Fig. 3 shows computed reaction profiles for the two possible acylations to form 2 (paths A and B) and β-hydride elimination to form 22 (path C). The three reaction profiles all start from the intermediate IN1, which is formed after initial oxidative addition of 2-iodobenzaldehyde, displacement of iodide by acetate and subsequent migratory insertion of 1.
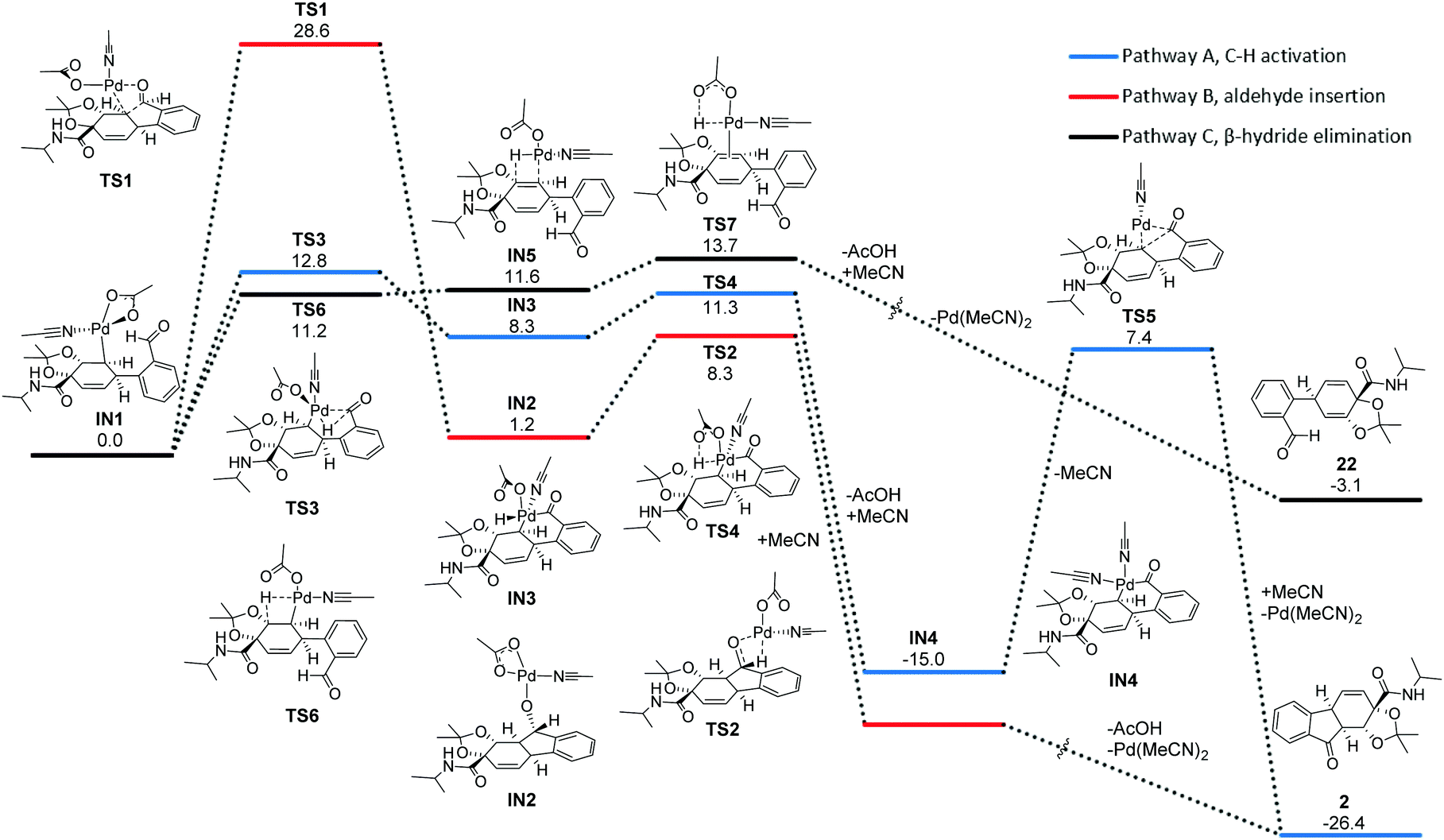 | ||
| Fig. 3 Gibbs free energy (1 M, 298.15 K, in kcal mol−1) reaction profile for the two proposed reaction paths and the competing β-hydride elimination. Geometry optimizations and frequency calculations were performed at the SMD-B3LYP-D3/def2-TZVPD//def2-SVPD level of theory. Energies are shown relative to IN1. Extended energy profiles for the aldehyde insertion and β-hydride elimination can be found in the ESI.† | ||
Our calculations predict path A, involving C–H activation, to be dominant, and clearly favoured over aldehyde insertion. The initial C–H activation step (TS3) has a barrier of 12.8 kcal mol−1 and is predicted to be the highest energy point along this reaction path. The second step of path A proceeds through deprotonation of the formed palladium(IV) complex (IN3) and loss of acetic acid through TS4, producing the palladium(II) species IN4. The IN4 intermediate is then predicted to undergo a reductive elimination through TS5 to form product 2. A possible alternative mechanism to pathway A is C–H activation through a concerted metalation-deprotonation,24 which has been suggested in related aryl acylations.13b,20a However, despite exhaustive efforts, no transition state for this type of mechanism could be located.
The aldehyde insertion (TS1) in pathway B computes as having a relatively high barrier of 28.6 kcal mol−1. A possible explanation for the high relative energy of TS1 is that the highly directional C(sp3)–Pd bond is almost completely broken in this transition state. Our modelling of this reaction shows that if aldehyde insertion would occur, then subsequent oxidation of the formed palladium alkoxide IN2 has a relatively low barrier. In other words, if IN2 is formed it could proceed to form product 2. Our calculations suggest that the rate determining step for path C is deprotonation of the palladium-hydride complex formed after the β-hydride elimination. This deprotonation (TS7) has a barrier of 13.7 kcal mol−1, which is ∼1 kcal mol−1 higher than the barrier for C–H activation. A predicted lower reaction rate for β-hydride elimination (path C) agrees with our experiments, where no arylation product 22 was observed. The backwards barrier for the alkene insertion forming IN1 was calculated to 19.1 kcal mol−1, making the formation of IN1 irreversible (ESI† Part 2). The migratory insertion transition state contains a hydrogen bond between the amide and the aldehyde, indicating that the amide can help facilitate the reaction with ortho aryl halides.
In summary, we have presented a palladium-catalyzed stereoselective domino arylation-cyclization reaction of enzymatically derived dienes with 2-iodobenzaldehydes. The combination of enzyme- and transition metal catalysis allows for introduction of a high degree of molecular complexity in just a few steps, from two simple aromatic precursors, i.e. benzoic acid and a 2-iodobenzaldehyde. The reaction proceeds in good yields and tolerates a range of aryl iodides, as well as some variation in terms of the diene substrate. In total, 12 homochiral products are reported with yields ranging from 14–74%. Quantum chemical calculations suggest that the cyclization is a result of a C–H activation that follows the initial Heck-type carbopalladation. The C–H activation is competitive despite the presence of a pendent β-hydrogen. This cyclization is, to our knowledge, the first example of a cyclative Heck-type alkene carbopalladation-acylation domino reaction.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Z. B. Yuan, Y. Y. Zeng, Z. W. Feng, Z. Guan, A. J. Lin and H. Q. Yao, Nat. Commun., 2020, 11, 2544 CrossRef CAS PubMed; (b) C. C. Wang, W. Y. Zhao, X. Q. Wu, J. P. Qu and Y. F. Chen, Adv. Synth. Catal., 2020, 362, 4996–5001 CrossRef CAS; (c) Y. Y. Ping, Y. X. Li, J. P. Zhu and W. Q. Kong, Angew. Chem., Int. Ed., 2019, 58, 1562–1573 CrossRef CAS PubMed; (d) J. X. Li, S. R. Yang, W. Q. Wu and H. F. Jiang, Chem. – Asian J., 2019, 14, 4114–4128 CrossRef CAS PubMed; (e) J. Biemolt and E. Ruijter, Adv. Synth. Catal., 2018, 360, 3821–3871 CrossRef CAS; (f) Q. S. Gu and D. Yang, Angew. Chem., Int. Ed., 2017, 56, 5886–5889 CrossRef CAS PubMed.
- (a) Z. Wang, Org. Biomol. Chem., 2020, 18, 4354–4370 RSC; (b) L. Xu, C. Wang, Z. W. Gao and Y. M. Zhao, J. Am. Chem. Soc., 2018, 140, 5653–5658 CrossRef CAS PubMed; (c) H. Ohno and S. Inuki, Synthesis, 2018, 700–710 CrossRef CAS.
- F. Ye, Y. Ge, A. Spannenberg, H. Neumann and M. Beller, Nat. Commun., 2020, 11, 5383 CrossRef CAS PubMed.
- W. H. Lin, J. M. Fang and Y. S. Cheng, Phytochemistry, 1995, 40, 871–873 CrossRef CAS.
- S. G. Cao, L. Ross, G. Tamayo and J. Clardy, Org. Lett., 2010, 12, 4661–4663 CrossRef CAS PubMed.
- C. M. Woo, L. Lu, S. L. Gholap, D. R. Smith and S. B. Herzon, J. Am. Chem. Soc., 2010, 132, 2540–2541 CrossRef CAS PubMed.
- (a) S. H. Gao, Q. L. Wang, L. J. S. Huang, L. Lum and C. Chen, J. Am. Chem. Soc., 2010, 132, 371–383 CrossRef CAS PubMed; (b) X. B. Liao, L. M. Stanley and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2088–2091 CrossRef CAS PubMed; (c) C. M. Woo, S. L. Gholap, L. Lu, M. Kaneko, Z. W. Li, P. C. Ravikumar and S. B. Herzon, J. Am. Chem. Soc., 2012, 134, 17262–17273 CrossRef CAS PubMed.
- J. W. Ruan, J. A. Iggo and J. L. Xiao, Org. Lett., 2011, 13, 268–271 CrossRef CAS PubMed.
- Y. Zhang, H. C. Shen, Y. Y. Li, Y. S. Huang, Z. Y. Han and X. Wu, Chem. Commun., 2019, 55, 3769–3772 RSC.
- N. Trongsiriwat, M. Y. Li, A. Pascual-Escudero, B. Yucel and P. J. Walsh, Adv. Synth. Catal., 2019, 361, 502–509 CrossRef CAS.
- For a recent report using organocatalysis, see: T. Shu, S. Li, X. Y. Chen, Q. Liu, C. von Essen, K. Rissanen and D. Enders, Chem. Commun., 2018, 54, 7661–7664 RSC.
- P. Álvarez-Bercedo, A. Flores-Gaspar, A. Correa and R. Martin, J. Am. Chem. Soc., 2010, 132, 466–467 CrossRef PubMed.
- (a) D. Solé, F. Mariani, I. Fernández and M. A. Sierra, J. Org. Chem., 2012, 77, 10272–10284 CrossRef PubMed; (b) D. Solé, F. Mariani and I. Fernandez, Adv. Synth. Catal., 2014, 356, 3237–3243 CrossRef; (c) D. Solé, F. Mariani and I. Fernandez, Eur. J. Org. Chem., 2015, 3935–3942 CrossRef.
- H. Viana, C. S. Marques, C. A. Correia, K. Gilmore, L. Galvao, L. Vieira, P. H. Seeberger and A. J. Burke, ChemistrySelect, 2018, 3, 11333–11338 CrossRef CAS.
- J. M. Liu, W. W. Song, Y. Y. Yue, R. Liu, H. Yi, K. L. Zhuo and A. W. Lei, Chem. Commun., 2015, 51, 17576–17579 RSC.
- (a) W. Tao, L. J. Silverberg, A. L. Rheingold and R. F. Heck, Organometallics, 1989, 8, 2550–2559 CrossRef CAS; (b) J. Vicente, J.-A. Abad and J. Gil-Rubio, Organometallics, 1996, 15, 3509–3519 CrossRef CAS; (c) R. C. Larock, M. J. Doty and S. Cacchi, J. Org. Chem., 1993, 58, 4579–4583 CrossRef CAS; (d) R. C. Larock, in Palladium in Organic Synthesis, ed. J. Tsuji, Springer, Berlin, Heidelberg, 2005, vol. 14, pp. 147–182 Search PubMed; (e) X. X. Zhang and R. C. Larock, Org. Lett., 2005, 7, 3973–3976 CrossRef CAS PubMed; (f) J. P. Waldo, X. X. Zhang, F. Shi and R. C. Larock, J. Org. Chem., 2008, 73, 6679–6685 CrossRef CAS PubMed; (g) V. Gevorgyan, L. G. Quan and Y. Yamamoto, Tetrahedron Lett., 1999, 40, 4089–4092 CrossRef CAS.
- (a) P. Shukla and C. H. Cheng, Org. Lett., 2006, 8, 2867–2869 CrossRef CAS PubMed; (b) Y. T. Chen, Z. T. Ding, Y. M. Wang, W. F. Liu and W. Q. Kong, Angew. Chem., Int. Ed., 2021, 60, 5273–5278 CrossRef CAS PubMed.
- (a) S. E. Lewis, Chem. Commun., 2014, 50, 2821–2830 RSC; (b) S. E. Lewis, in Asymmetric Desymmetrization Reactions, ed. S.-L. You, Wiley VCH Verlag GmbH & Co. KGaA., Weinheim, 2016 Search PubMed.
- A. J. Paterson, P. Dunås, M. Rahm, P. O. Norrby, G. Kociok-Köhn, S. E. Lewis and N. Kann, Org. Lett., 2020, 22, 2464–2469 CrossRef CAS PubMed.
- (a) A. Flores-Gaspar, A. Gutierrez-Bonet and R. Martin, Org. Lett., 2012, 14, 5234–5237 CrossRef CAS PubMed; (b) C. D. Pan, X. F. Jia and J. Cheng, Synthesis, 2012, 677–685 CAS.
- D. Solé and I. Fernandez, Acc. Chem. Res., 2014, 47, 168–179 CrossRef PubMed.
- S. K. Meegalla, N. J. Taylor and R. Rodrigo, J. Org. Chem., 1992, 57, 2422–2427 CrossRef CAS.
- M. J. Frisch et al. Gaussian 16 Rev. C.01, Wallingford, CT, 2016.
- S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2043870 (21). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc02160e |
| This journal is © The Royal Society of Chemistry 2021 |