DOI:
10.1039/D1CC02112E
(Feature Article)
Chem. Commun., 2021,
57, 6111-6120
Recent advances in acyl radical enabled reactions between aldehydes and alkenes
Received
21st April 2021
, Accepted 25th May 2021
First published on 25th May 2021
Abstract
Radical-mediated functionalization of alkenes has been emerging as an elegant and straightforward protocol to increase molecule complexity. Moreover, the abstraction of a hydrogen atom from aldehydes to afford acyl radicals has evolved as a rising star due to its high atom-economy and the ready availability of aldehydes. Considering the great influence and synthetic potential of acyl radical enabled reactions between aldehydes and alkenes, we provide a summary of the state of the art in this field with a specific emphasis on the working models and corresponding mechanisms. The discussion is divided according to the kind of alkenes and reaction type.
1 Introduction
Alkenes are one of the most valuable building blocks and have found wide applications in chemical synthesis, pharmaceutical discovery, and material science.1 Hence, the development of efficient methods for the functionalization of alkenes has become an important and well-studied research topic in organic synthesis. In particular, radical-mediated functionalization of alkenes has emerged as an elegant and straightforward protocol to increase molecule complexity with features of high step- and atom-economic efficiency, high reactivity, and excellent functional group tolerance.2
Since the beginning of radical chemistry, the formation and application of acyl radicals in a controlled and efficient manner has been at the heart of organic synthesis.3 Among such strategies, the generation of alkyl radicals and their subsequent carbonylation has become a prominent tool.4 In addition, the use of acyl selenides together with a radical initiator has also been developed as a complementary approach to directly generating acyl radicals.5 As an alternative to these classical synthetic methods, the photochemical cleavage of the RC(O)–X (X = Cl, TeR, POR2, CH(OH)R, CH(NH2)R, OH and so on) bond has emerged as a powerful platform to deliver acyl radicals.6 Peroxide-promoted decarboxylation of α-keto acids is also a viable alternative to obtain acyl radicals.7 Recently, the abstraction of a hydrogen atom from aldehydes to afford acyl radicals has evolved as a rising star in this field due to its high atom-economy and the ready availability of aldehydes.3b,8
However, to the best of our knowledge, to date, there is still no exclusive review devoted to acyl radical enabled reactions between aldehydes and alkenes. Hence, we will endeavor to highlight the recent advances in this topic. Specific emphasis has been placed on working models and corresponding mechanisms. The following discussion is thus organized based on the kind of alkenes and reaction type, which includes hydroacylation of simple alkenes and difunctionalization of alkenes. The authors hope that this review will inspire further development of reactions in this promising and interesting area.
2 Hydroacylation of simple alkenes
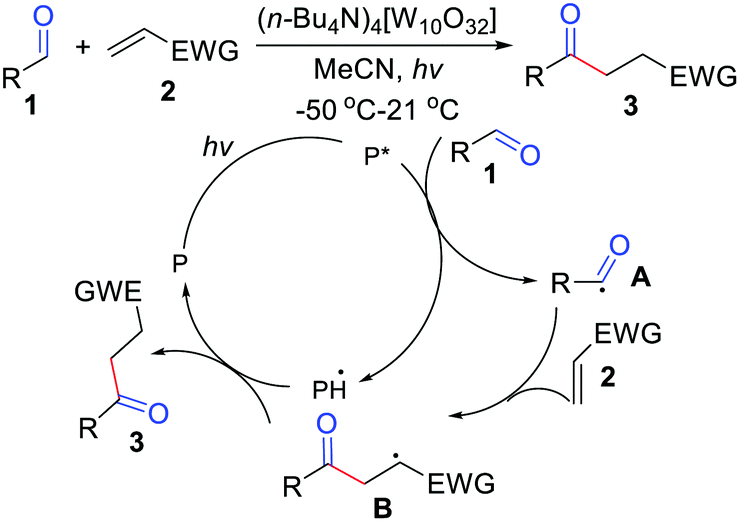
Initial efforts in hydroacylation of simple alkenes via decatungstate-photocatalyzed activation of aldehydes were made by Fagnoni and co-workers (Scheme 1).9 The reaction involved the use of tetra-butyl ammonium decatungstate (TBADT) at 2 mol% amount and did not require the use of foul-smelling auxiliaries. After establishing a suitable system for the production of acyl radicals, a variety of electron-poor alkenes were subjected to optimal reaction conditions and afforded the hydroacylation products of asymmetrical ketones in modest yields. The process involves the generation of an acyl radical through hydrogen abstraction from aldehydes, followed by radical addition to the alkene to give the radical intermediate B. Then, it abstracts a hydrogen to release the final product. Inspired by the work mentioned above, other groups have also reported hydroacylation of simple alkenes using aldehydes.10
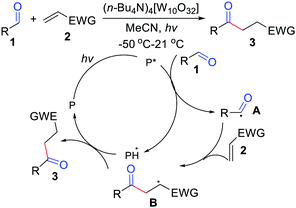 |
| | Scheme 1 Decatungstate-photocatalyzed activation of aldehydes with electron-poor alkenes. | |
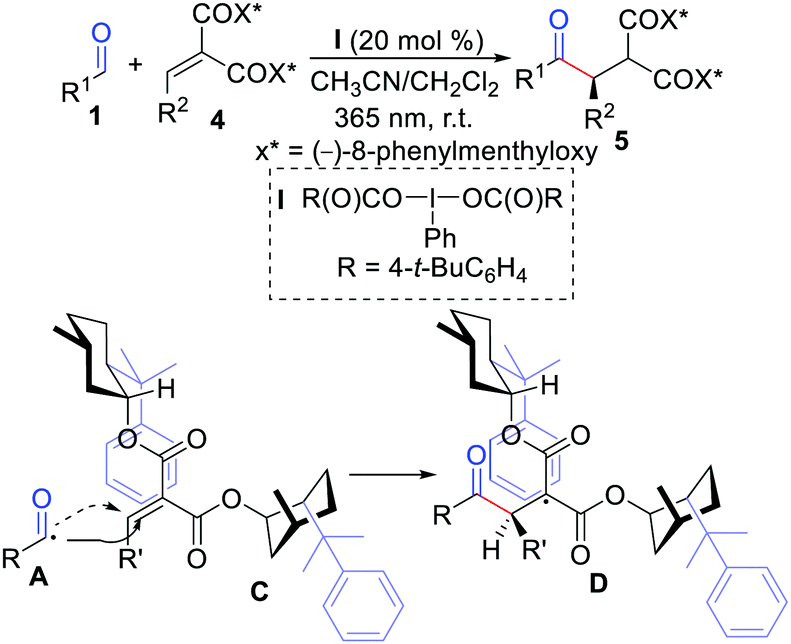
The first example of diastereoselective radical hydroacylation of alkenes was described by Maruoka and co-workers in 2016 (Scheme 2).11 The reaction was initiated by the combination of hypervalent iodine(III) with UV-light irradiation under metal-free conditions. They also proposed that a transition state model for this transformation is a radical addition step, and effective shielding of one diastereotopic face of alkylide-nemalonates was realized by the phenyl group of one chiral auxiliary.
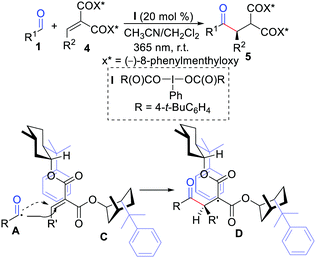 |
| | Scheme 2 Diastereoselective radical hydroacylation of alkenes. | |
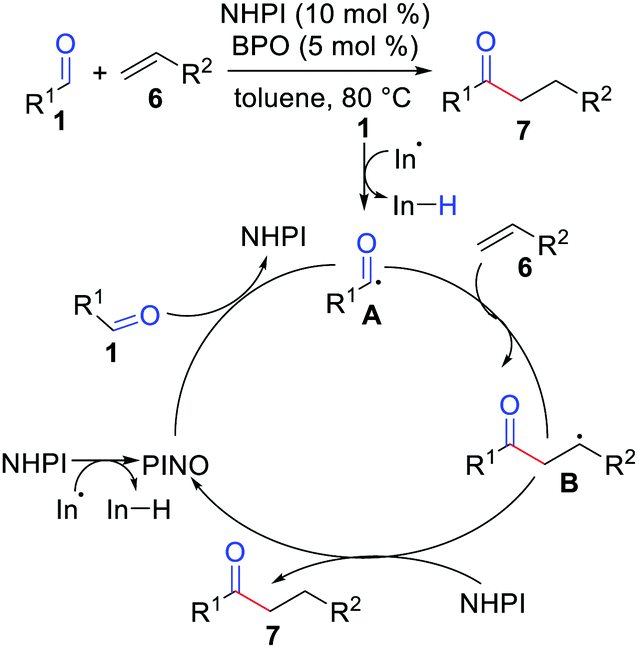
Ishii and co-workers in 2001 reported the hydroacylation of simple alkenes with aldehydes with the use of a polarity-reversal catalyst (Scheme 3).12 In this report, they tried several N-hydroxyphthalimide (NHPI) derivatives and N-hydroxysuccinimide (NHSI) as catalysts, but the combination of NHPI with dibenzoyl peroxide (BPO) was found to be most suitable for this particular method.
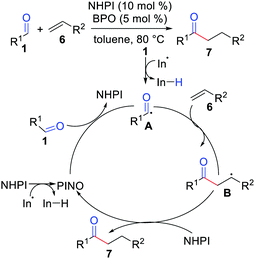 |
| | Scheme 3 Hydroacylation of simple alkenes with aldehydes by the use of a polarity-reversal catalyst. | |
3 Difunctionalization of alkenes
3.1 Aldehyde-mediated coupling of an acyl radical with simple alkenes
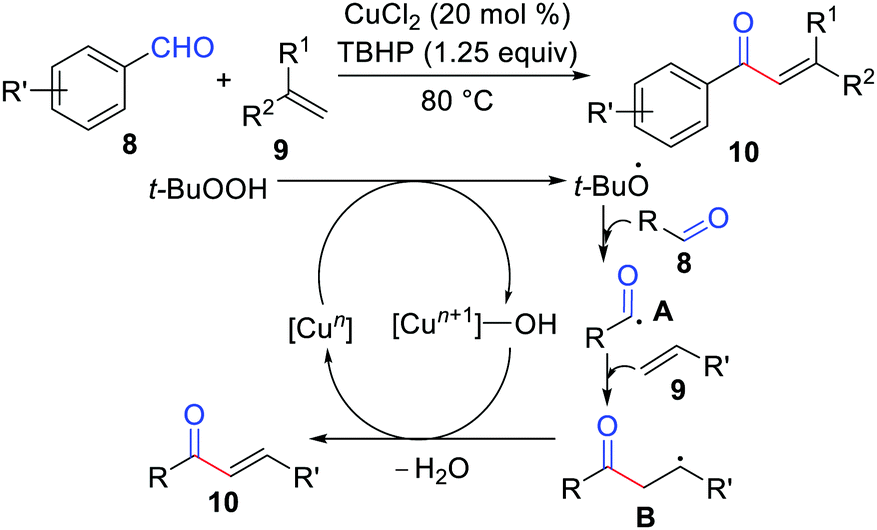
Oxidative coupling has emerged as one of the most popular and powerful methodologies because it avoids the use of halides (or halide equivalents) and organometallic reagents.13 In 2013, Lei's group developed the first oxidative coupling of alkenes with aldehydes for the synthesis of α,β-unsaturated ketones (Scheme 4).14 The mechanistic studies showed that this reaction likely proceeded by a single-electron transfer (SET), and the important benzylic radical intermediate B underwent oxidation by copper species and then deprotonation to produce the final coupling product. Recently, Zhao, Zhu, Loh, and co-workers also proposed an oxidative coupling of alkenes with aldehydes via a photoredox catalysis method.15
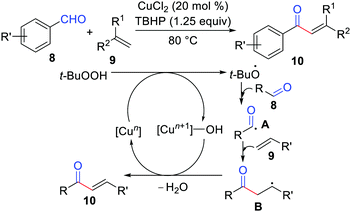 |
| | Scheme 4 Oxidative coupling of alkenes with aldehydes for the synthesis of α,β-unsaturated ketones. | |
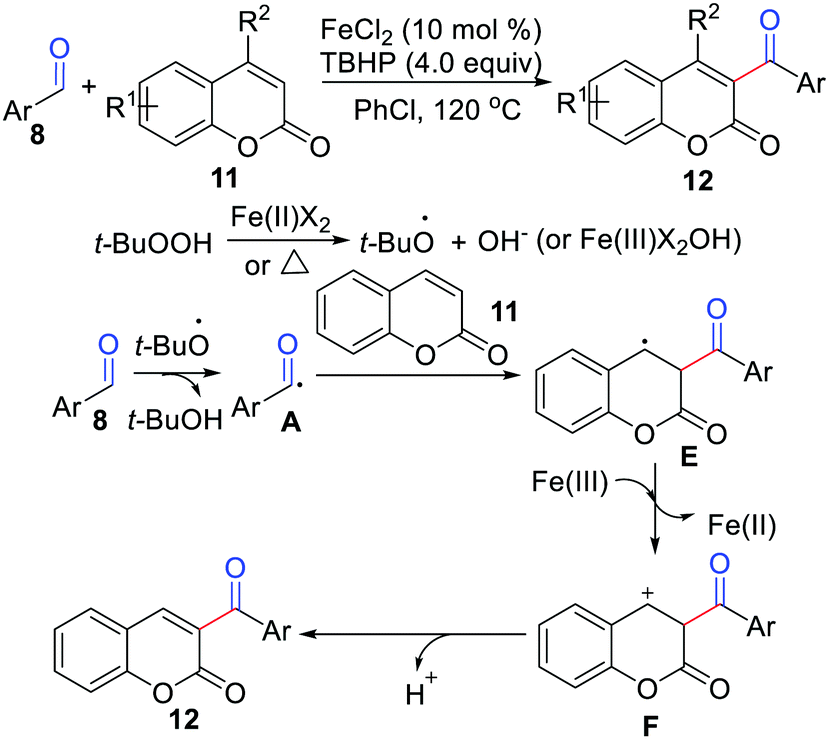
In 2015, Yuan, Mao, Qu, and co-workers developed a novel iron-catalyzed protocol for the coupling of aromatic aldehydes with coumarins (Scheme 5).16 The features of this method include high efficiency, wide functional group tolerance, and commercially available starting materials. They proposed a SET reaction process, which underwent a carbocation intermediate for the formation of the final product. Soon thereafter, Adib and co-workers further reported a metal-free coupling of aldehydes with coumarins in the presence of a K2S2O8/aliquat 336 system.17
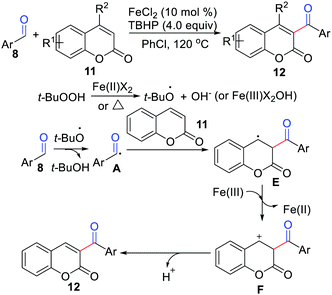 |
| | Scheme 5 Iron-catalyzed protocol for the coupling of aromatic aldehydes with coumarins. | |
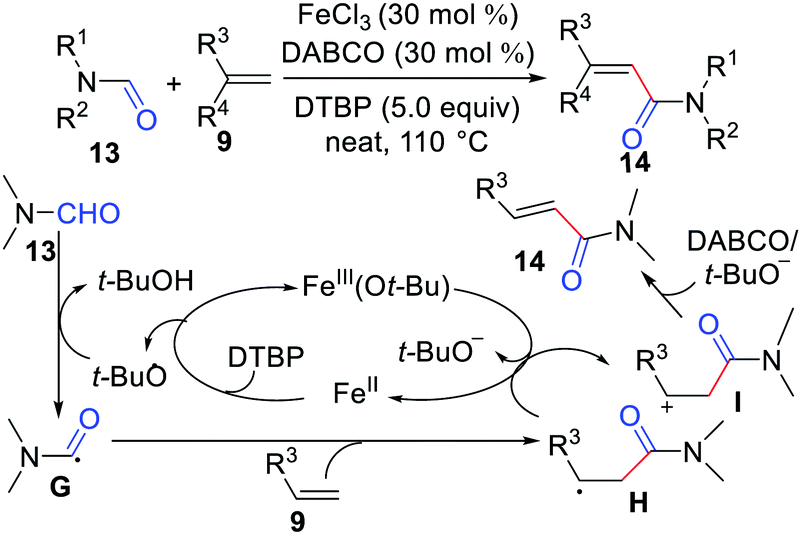
In 2014, Li's group achieved oxidative coupling of alkenes with amides via carbonyl C(sp2)–H functionalization (Scheme 6).18 In this reaction, FeCl3 was the catalyst, 1,4-diazabicyclo[2.2.2]octane (DABCO) was the base, and di-tert-butyl peroxide (DTBP) was the oxidant. The mechanism involves the SET between alkyl radical H and Fe3+(Ot-Bu) to form alkyl cation I, followed by β-H elimination affording the desired product as shown in Scheme 6.
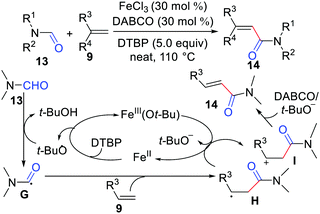 |
| | Scheme 6 Oxidative coupling of alkenes with amides via carbonyl C(sp2)–H functionalization. | |
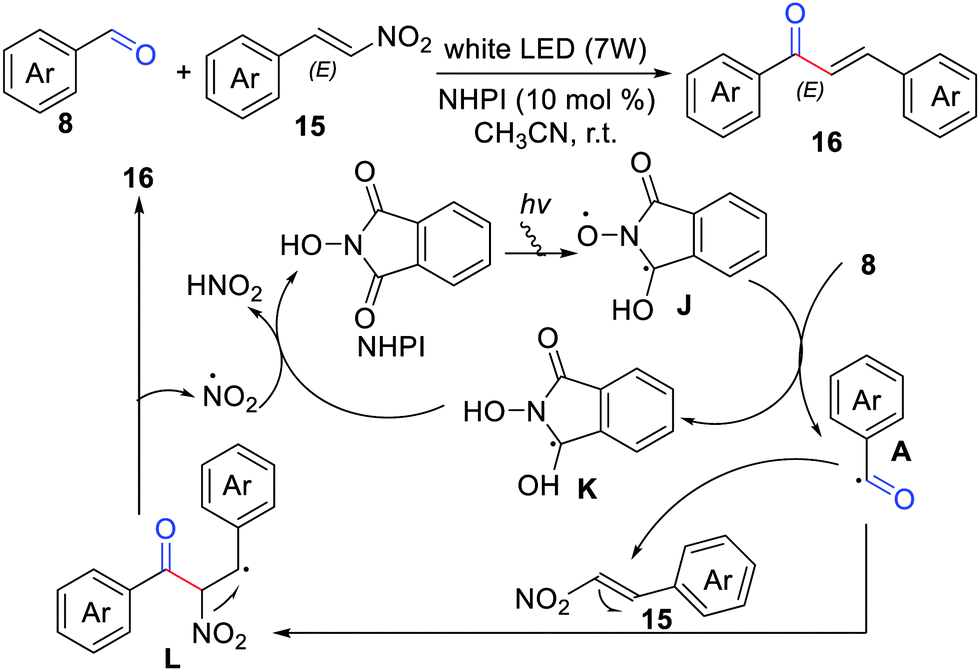
In 2017, Yadav and co-workers developed the coupling of aldehydes with β-nitrostyrenes towards chalcones, by means of N-hydroxyphthalimide (NHPI) as a reusable organophotocatalyst and acetonitrile as an acceptable green solvent (Scheme 7).19 A possible reaction mechanism has been suggested by the authors considering that the benzylic radical L easily eliminates the stable NO2 radical to form the desired product.
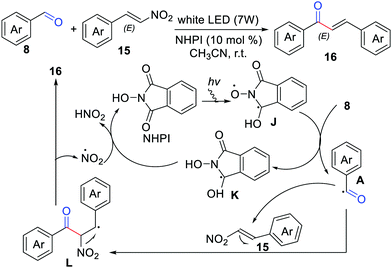 |
| | Scheme 7 The coupling of aldehydes with β-nitrostyrenes. | |
3.2 Three-components reaction of aldehydes with alkenes
α,β-Epoxy ketones are a class of valuable intermediates and precursors in organic chemistry, and they can be easily converted into numerous useful products such as α- and β-carbonyls, α,β-epoxy alcohols, 1,3-diols and so on.20
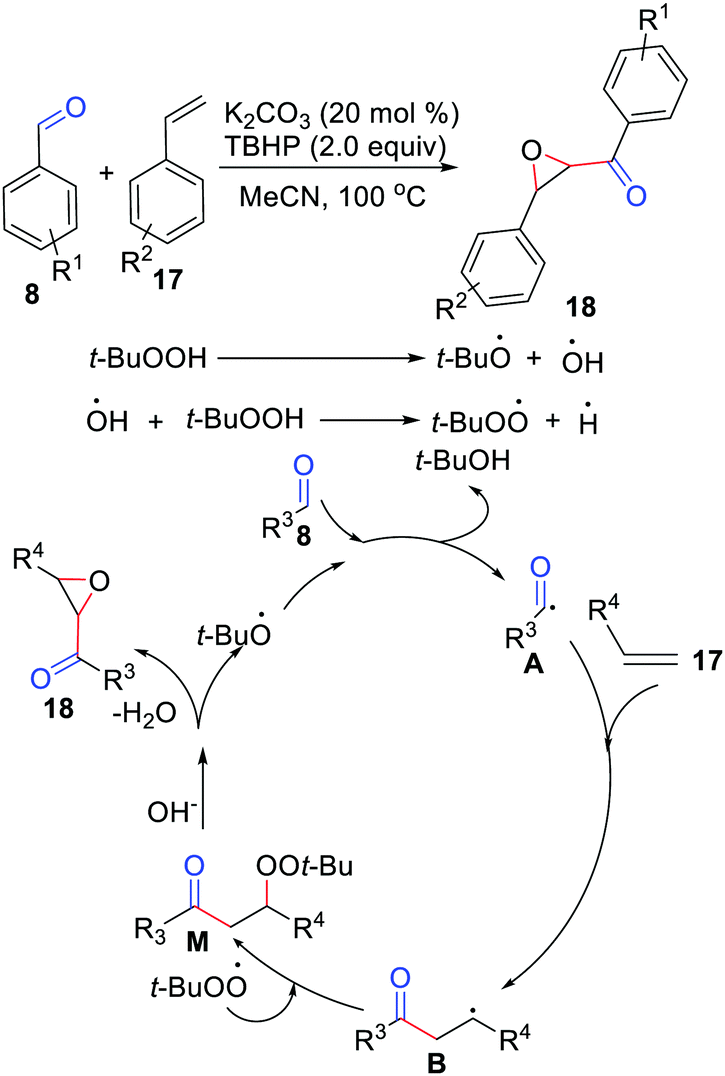
The difunctionalization of simple alkenes for the synthesis of α,β-epoxy ketones via oxidative coupling with aldehydes under transition metal-free conditions had been described by the Lu group in 2014 (Scheme 8).21 Various aldehydes were found to be tolerated, both aryl and alkyl aldehydes ran well in this reaction. An investigation of the mechanism showed that the reaction involved the formation of an acyl radical, radical addition, radical coupling, and the elimination of t-BuOH.
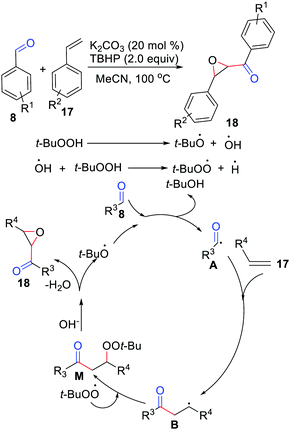 |
| | Scheme 8 The difunctionalization of simple alkenes for the synthesis of α,β-epoxy ketones. | |
Next, Li and co-workers reported an elegant one-pot synthesis of α,β-epoxy ketones using 10 mol% t-BuOK and 2.0 equiv tert-butyl hydroperoxide (TBHP). (Scheme 9).22 This reaction proceeds through a new tandem C–H/alkene functionalization step that occurs through an oxidative radical pathway. Subsequently, the Wang, Siva, and Jr group independently demonstrated the synthesis of α,β-epoxy ketones through a domino reaction between alkenes and aldehydes via a visible light-driven strategy.23
 |
| | Scheme 9 One-pot synthesis of α,β-epoxy ketones. | |
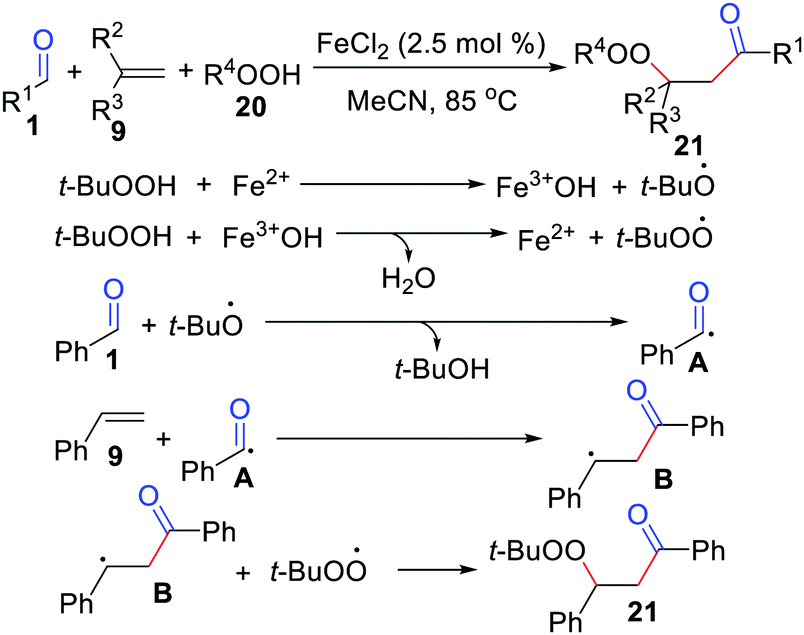
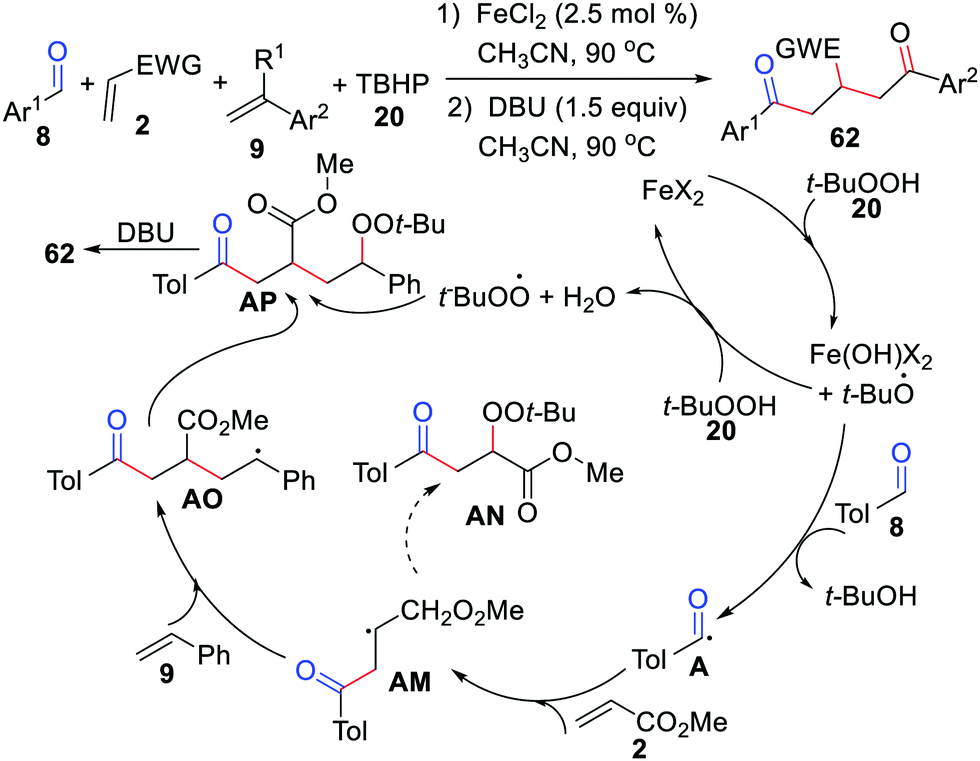
Oxidative difunctionalization of alkenes, which involves the installation of two substituents across a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, has attracted significant attention as an unprecedented method that allows for the buildup of molecular complexity in a single procedure.24 In 2011, the Li group presented an FeCl2-catalyzed acylation/peroxidation of alkenes with aldehydes and hydroperoxides for the preparation of β-peroxy ketones (Scheme 10).25 In this case, α-substituted alkenes, 2-vinylnaphthalene, 4-vinylpridine, and 1,3-butadiene were suitable for this transformation. Remarkably, the method can also be applied to the synthesis of α-carbonyl epoxides through a one-pot process by adding 10 mol% 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to the reaction mixture after the completion of the acylation/peroxidation reaction. The acyl radical addition to the CC bond of the alkene followed by alkylperoxy radical coupling leads to the expected product 21. Moreover, the groups of Li26 and Wang27 independently exploited acylation/peroxidation of alkenes using iron or dirhodium.
C double bond, has attracted significant attention as an unprecedented method that allows for the buildup of molecular complexity in a single procedure.24 In 2011, the Li group presented an FeCl2-catalyzed acylation/peroxidation of alkenes with aldehydes and hydroperoxides for the preparation of β-peroxy ketones (Scheme 10).25 In this case, α-substituted alkenes, 2-vinylnaphthalene, 4-vinylpridine, and 1,3-butadiene were suitable for this transformation. Remarkably, the method can also be applied to the synthesis of α-carbonyl epoxides through a one-pot process by adding 10 mol% 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to the reaction mixture after the completion of the acylation/peroxidation reaction. The acyl radical addition to the CC bond of the alkene followed by alkylperoxy radical coupling leads to the expected product 21. Moreover, the groups of Li26 and Wang27 independently exploited acylation/peroxidation of alkenes using iron or dirhodium.
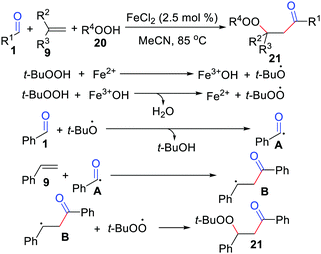 |
| | Scheme 10 FeCl2-Catalyzed acylation/peroxidation of alkenes with aldehydes and hydroperoxides. | |
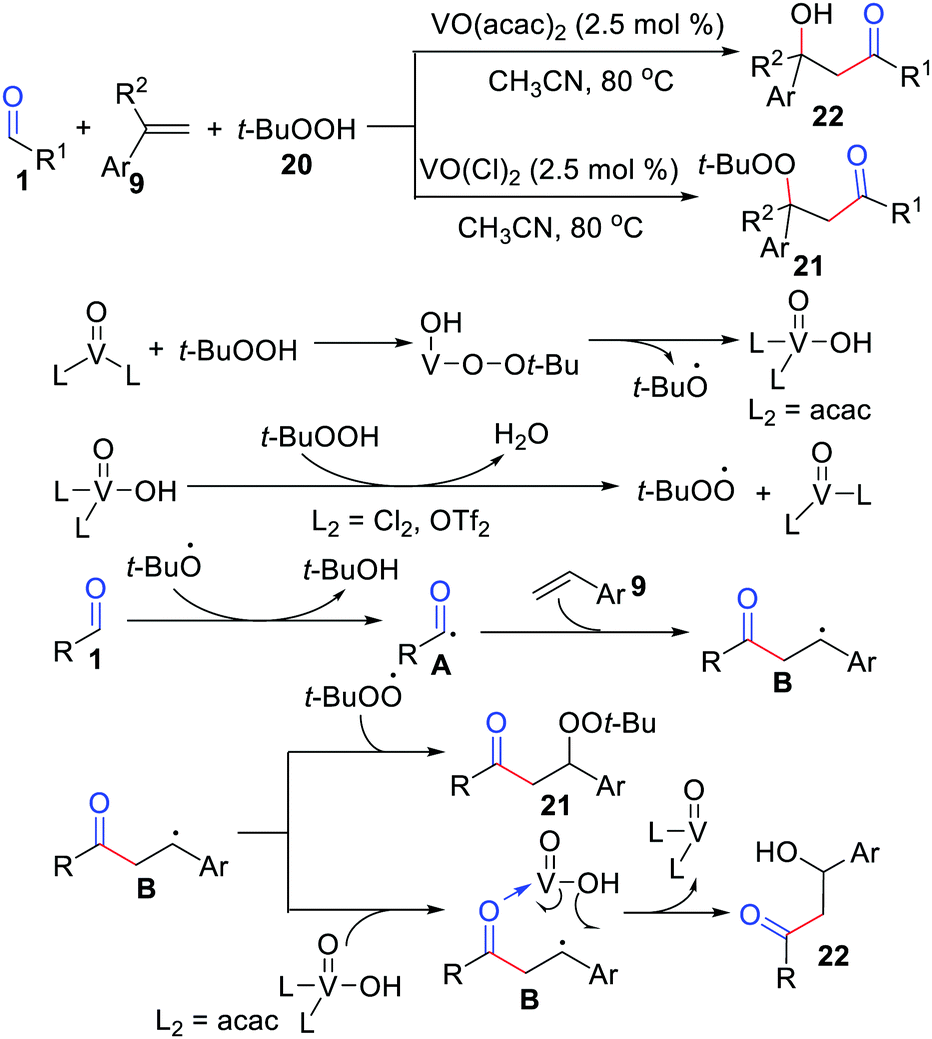
Subsequently, Weng, Chen, and co-workers further demonstrated complementary acylation/peroxidation and acylation/hydroxylation of alkenes by simply adjusting the ligand environment of a vanadyl center (Scheme 11).28 This method was attractive and selectively afforded β-peroxyketones or β-hydroxy using the same set of coupling partners. The acylation/hydroxylation may proceed through the direct coupling of vanadyl(V) hydroxide with benzylic radical B.
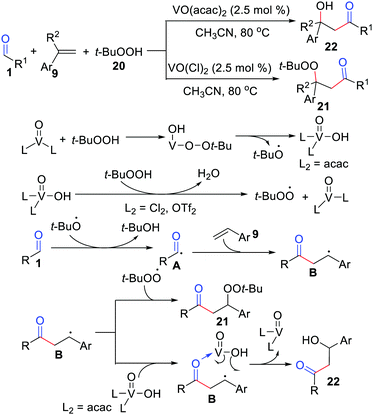 |
| | Scheme 11 The complementary acylation/peroxidation and acylation/hydroxylation of alkenes. | |
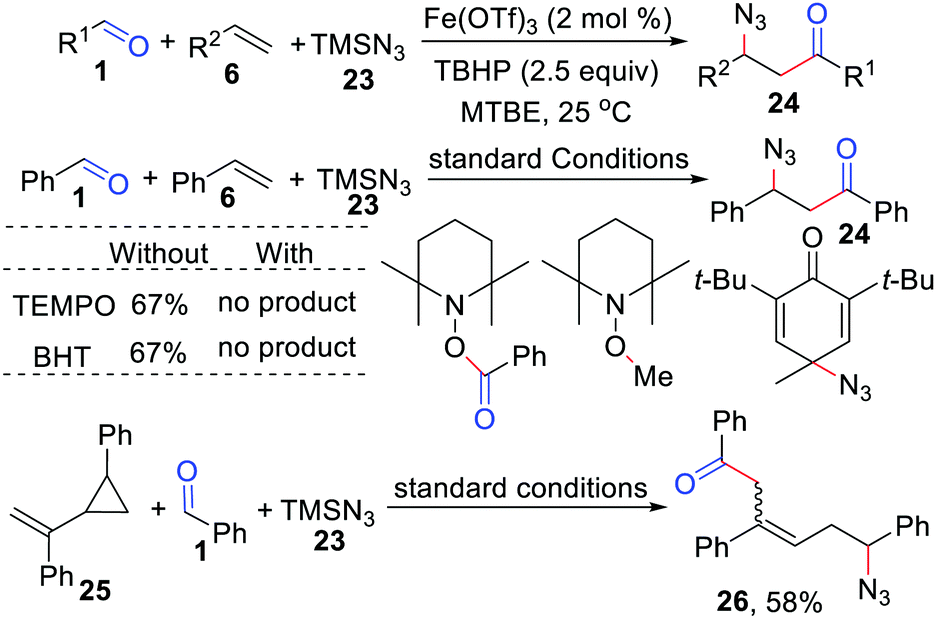
In 2019, Bao and co-workers disclosed an iron-catalyzed acylation/azidation of alkenes by using TMSN3 as the azido source and TBHP as the initiator, providing a series of valuable unsymmetrical β-azido ketones in moderate to good yield (Scheme 12).29 The obtained β-azido ketones can be easily transformed into γ-aminol, γ-azido alcohol, β-azido oxime, β-azido ester, or triazoles. Radical trapping experiments confirmed that this reaction involves the azido and acyl radical processes.
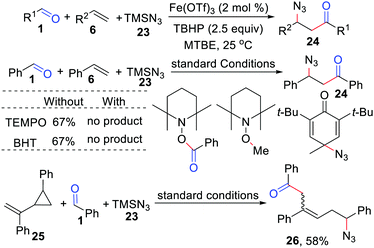 |
| | Scheme 12 Iron-catalyzed acylation/azidation of alkenes. | |
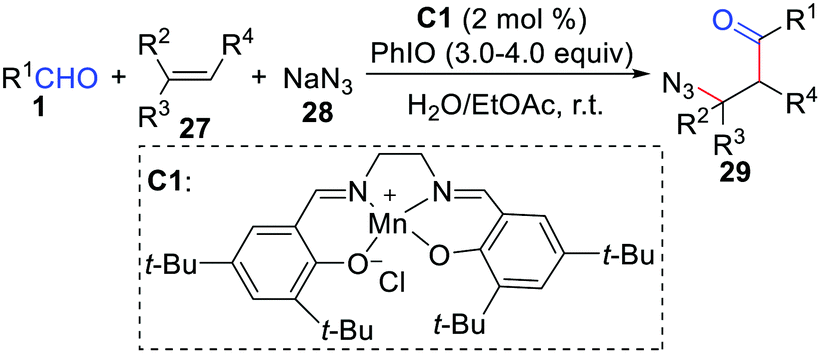
Generally, (salen)Mn(III) in conjunction with iodosobenzene is a novel catalysis system for epoxidation of alkenes. However, the Wang group developed a (salen)Mn(III)-catalyzed chemoselective acylation/azidation of alkenes by using NaN3 as the azido source (Scheme 13).30 The key to this protocol is the rational application of the different reactivities of the oxomanganese(V) species, which is capable of abstracting a hydrogen atom from a substrate C–H bond.
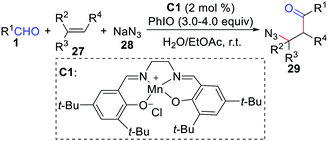 |
| | Scheme 13 (Salen)Mn(III)-catalyzed chemoselective acylation/azidation of alkenes. | |
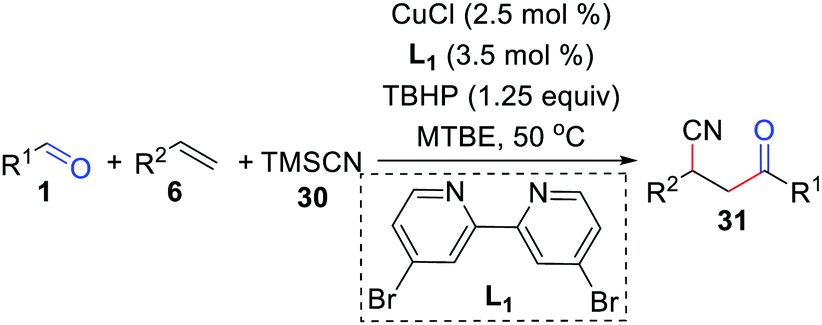
In 2019, Bao and co-workers further achieved a copper-catalyzed acyl/cyanation of alkenes using TBHP as an initiator (Scheme 14).31 In this transformation, the radical pathway starts from a tert-butoxy radical and the dominant factor for the β-Me scission of a tert-butoxy radical is the metal species.
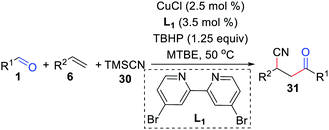 |
| | Scheme 14 Copper-catalyzed acyl/cyanation of alkenes. | |
3.3 Intermolecular cyclization of aldehydes with alkenes
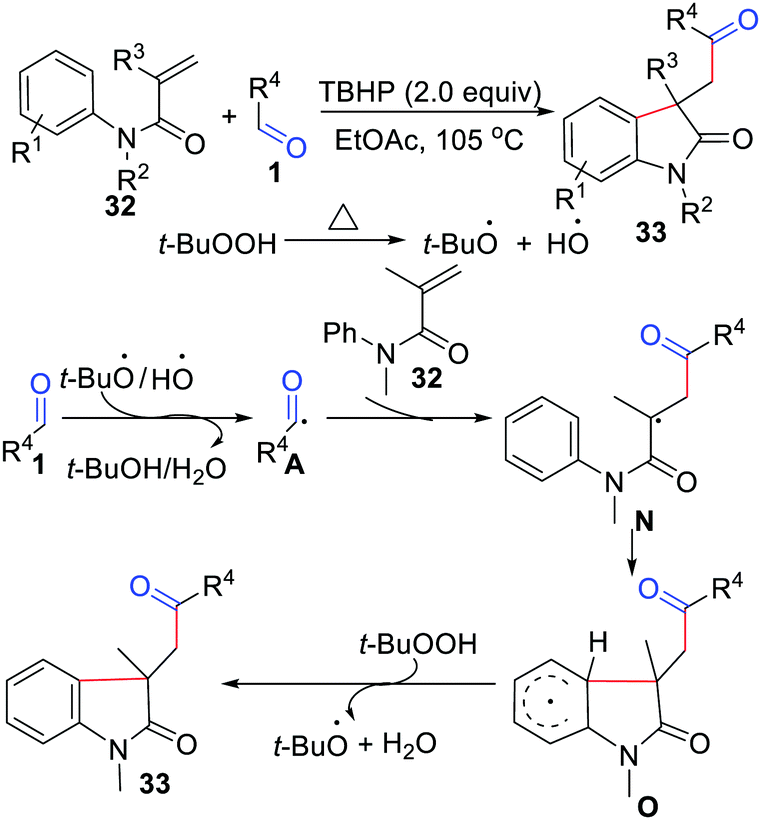
The first intermolecular acylation/cyclization of N-arylacrylamides was reported in 2013 by Song, Li, and co-workers, and it established a metal-free oxidative difunctionalization of activated alkenes with carbonyl C(sp2)–H bonds and aryl C(sp2)–H bonds (Scheme 15).32 In this reaction, TBHP and DTBP were exclusive oxidants, as traces of the target products were detected in other oxidants. Notably, the transformation showed an obvious steric hindrance effect and afforded a lower yield with ortho-substituted aryl aldehydes. They also proposed a possible mechanism, where an acyl radical was produced by TBHP. Then it reacted with the CC bond of N-arylacrylamides to generate radical intermediate N, which was further cyclized with an aryl ring to give radical intermediate O. Finally, the product was obtained via abstraction of an aryl hydrogen by TBHP. Since then, different variants involving acylation/cyclization of N-arylacrylamides with aldehydes have been developed.33
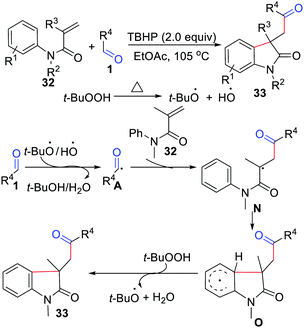 |
| | Scheme 15 Intermolecular acylation/cyclization of N-arylacrylamides. | |
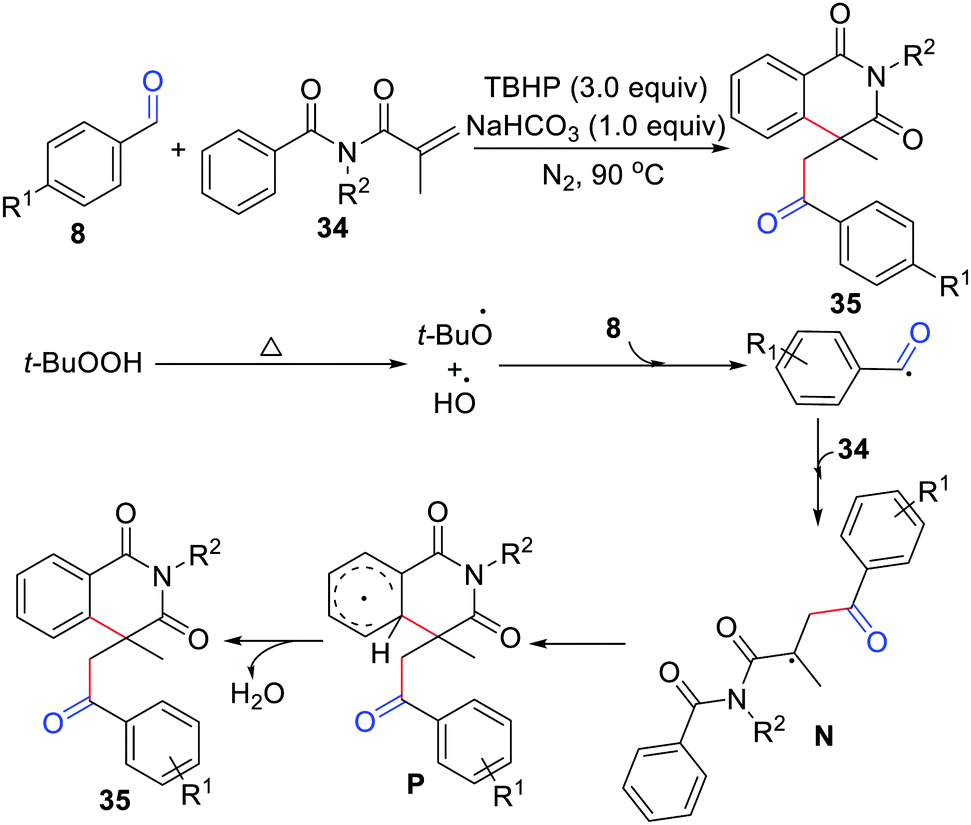
One year later, the Zhou group disclosed acylation/cyclization of N-alkyl-N-methacryloylbenzamide for the synthesis of isoquinoline-1,3(2H,4H)-diones (Scheme 16).34 In this method, TBHP was the oxidant, NaHCO3 was the base, and no metal catalysts or organic solvents were involved. In addition, the groups of Mai/Lu35 and Duan/Li36 independently exploited acylation/cyclization of N-alkyl-N-methacryloylbenzamide simply using K2S2O8 or DTBP as the oxidant.
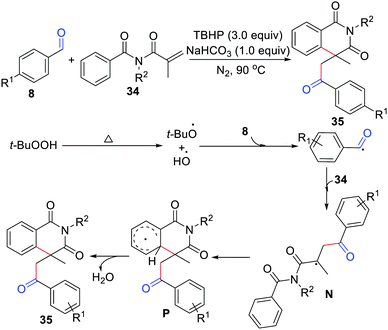 |
| | Scheme 16 Acylation/cyclization of N-alkyl-N-methacryloylbenzamide. | |
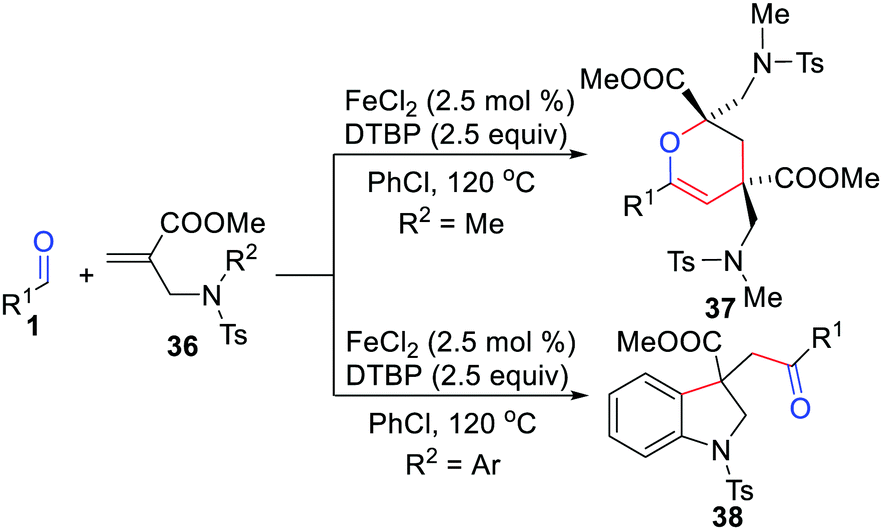
In 2015, Li's group expanded this cyclization protocol to N-allylamides for the rapid and efficient preparation of indolines and dihydropyrans using FeCl2/DTBP (Scheme 17).37 Notably, the substituents of tertiary amines play vital roles in facilitating the desired transformation as shown in Scheme 17. In 2020, Liang, Long, and co-workers reported a heat- or light-induced acylarylation of N-allylamides for the construction of 3-(α-acyl) indolines.38
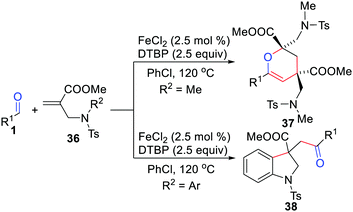 |
| | Scheme 17 Cyclization of N-allylamides for the preparation of indolines and dihydropyrans. | |
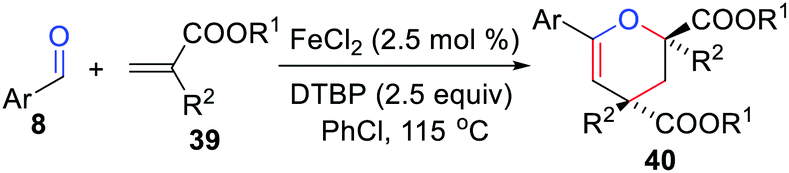
In 2015, Li and co-workers developed an efficient iron-catalyzed cyclization of aldehydes with alkenes for the synthesis of 3,4-dihydropyran derivatives (Scheme 18).39 Iron salts are an efficient and irreplaceable catalyst, as the target products were not detected in the absence of it. Soon after, the same group reported an iron-catalyzed addition cyclization reaction of aldehydes with alkenes.40
 |
| | Scheme 18 Iron-catalyzed cyclization of aldehydes with alkenes. | |
In the same year, the same group realized the iron-catalyzed acylation/oxygenation of terminal alkenes with aldehydes for the preparation of dihydrofurans bearing a quaternary carbon (Scheme 19).41 A control experiment suggested that high temperature (120 °C) was essential for this reaction, as no product was detected when the reaction was implemented at 100 °C.
 |
| | Scheme 19 Iron-catalyzed acylation/oxygenation of terminal alkenes for the preparation of dihydrofurans. | |
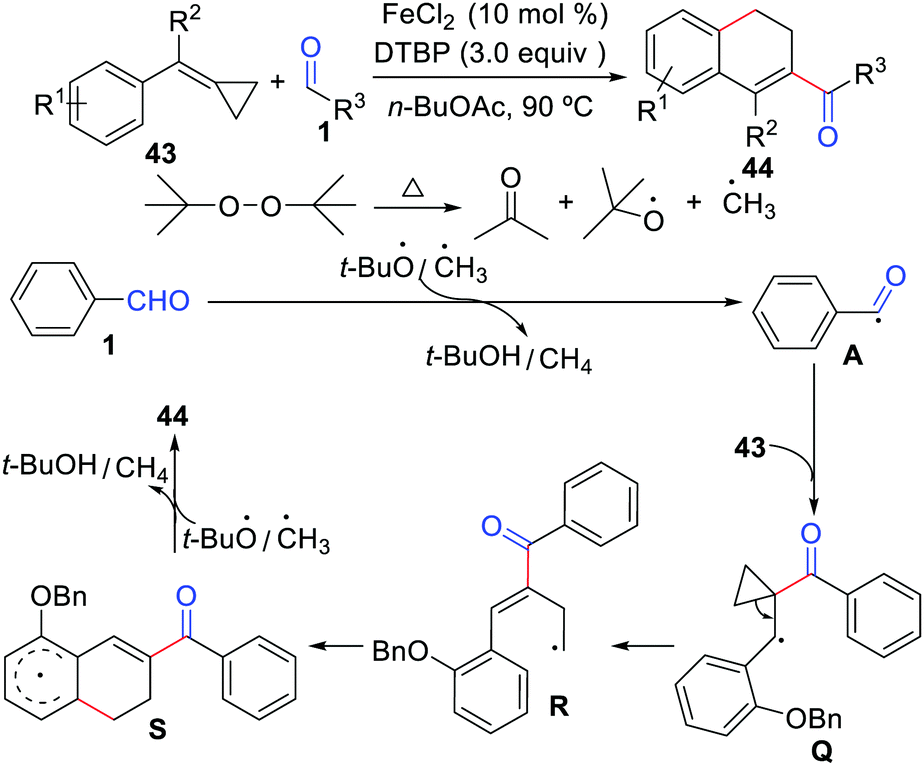
Despite the impressive advances made in the intermolecular cyclization of alkenes with aldehydes, employing methylenecyclopropanes as the partner in these reactions remains largely unexplored. Hence, Liu, Tang, and co-workers developed the first example of functionalization of methylenecyclopropanes with aldehydes for the formation of 2-acyl-3,4-dihydronaphthalenes using FeCl2 as the catalyst (Scheme 20).42 This cyclization reaction proceeds through an acyl radical addition, ring-opening, and intramolecular cyclization sequence.
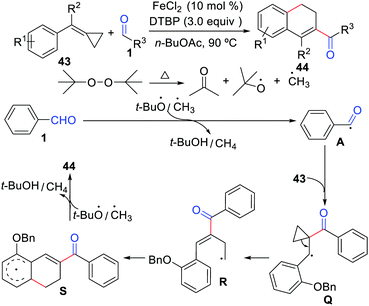 |
| | Scheme 20 Functionalization of methylenecyclopropanes with aldehydes. | |
3.4 Difunctionalization of aldehydes and unactivated alkenes under the strategy of functional group ipso-migration
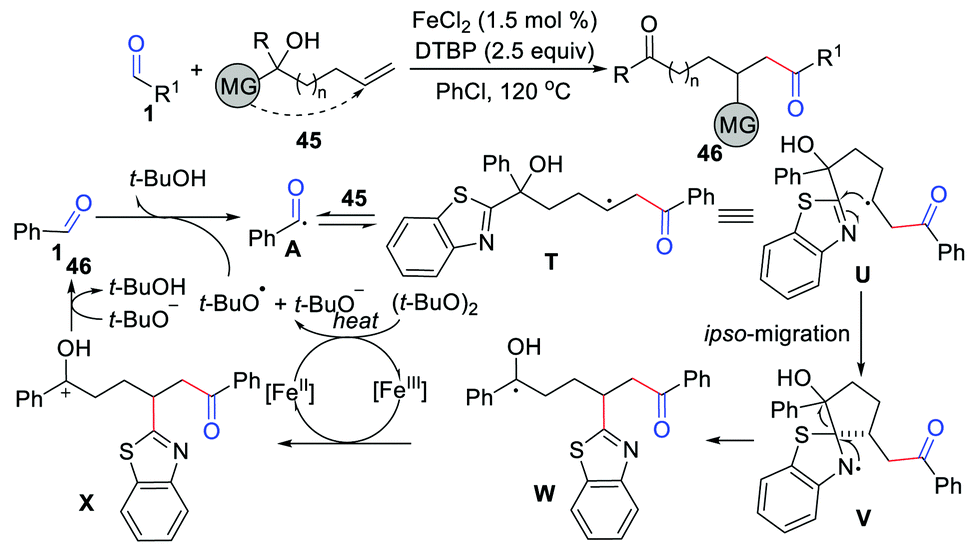
To further exploit the hydroacylation of unactivated alkenes with aldehydes, Lv, Li, and co-workers have disclosed an effective distal group ipso-migration strategy in 2020 (Scheme 21).43 This method overcomes the energy barrier and reversibility in the functionalization of unactivated alkenes with nucleophilic acyl radicals. The key ipso-migration step of this transformation is the β-carbonyl carbon radical U trapped by the CN bond of the alkene substrate to give the nitrogen radical intermediate V. Moreover, Yu's group reported a similar 1,2-acylarylation of allylic alcohols with aldehydes in 2017.44
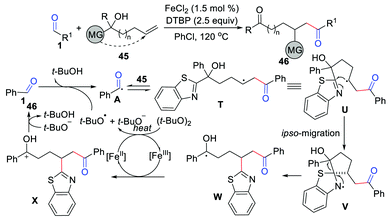 |
| | Scheme 21 The hydroacylation of unactivated alkenes with aldehydes. | |
3.5 Intramolecular cyclization of alkenes
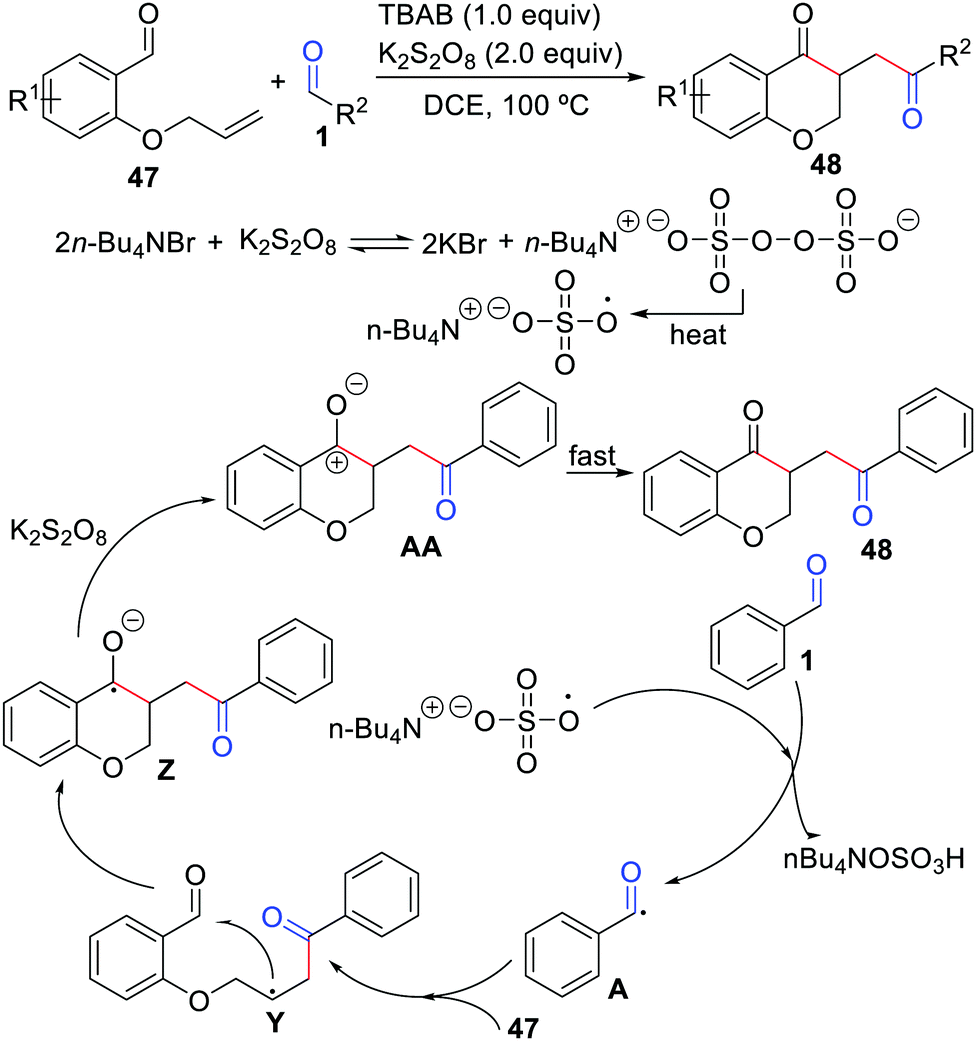
The first example of cyclization of 2-(allyloxy)arylaldehydes with aldehydes was described by Xiao, Mai, and co-workers in 2019 (Scheme 22).45 In the presence of tetra-n-butylammonium bromide (TBAB) and K2S2O8, a series of chroman-4-ones were produced in moderate to good yields. The strategy features easily available starting materials, no metals, and a step-economical manner. This reaction involves a benzyl cation intermediate AA, which could be transformed into the final product fast.
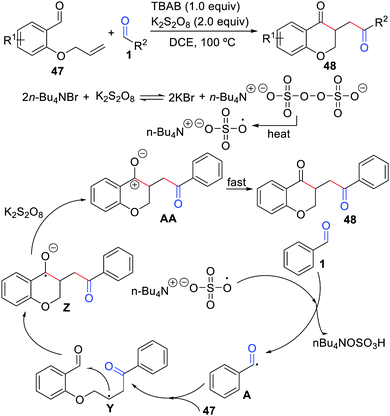 |
| | Scheme 22 Cyclization of 2-(allyloxy)arylaldehydes with aldehydes. | |
Lee et al. achieved the TBHP-promoted hydroacylation of 2-(allyloxy)benzaldehydes for the synthesis of 4-chromanones under solvent- and metal-free conditions (Scheme 23).46 The method proceeded through the production of an intramolecular acyl radical, followed by the reaction with a CC bond.
 |
| | Scheme 23 TBHP-promoted hydroacylation of 2-(allyloxy)benzaldehydes. | |
Hong disclosed a tandem oxidation/radical cyclization/epoxidation sequence for the preparation of spiroepoxy chroman-4-ones through a visible-light-enabled protocol in 2017 (Scheme 24).47 Optimization of the reaction conditions showed that with mediation by 1 mol% of Ru(bpy)3Cl2·6H2O, 5.0 equiv. of TBHP, and 3.0 equiv. of K2CO3 in i-PrOAc at room temperature, the desired product can be obtained in 73% yield after 24 h with irradiation from a blue LED.
 |
| | Scheme 24 Preparation of spiroepoxy chroman-4-ones. | |
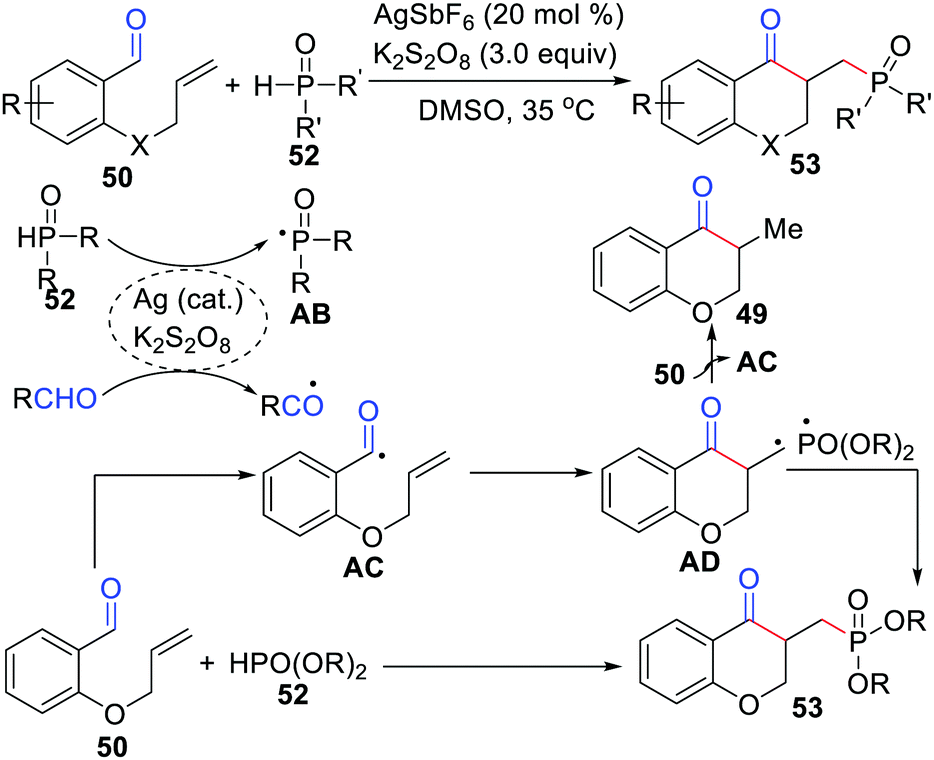
Li and co-workers reported an efficient and unique approach to synthesize phosphonate chroman-4-ones employing AgSbF6 as the catalyst and K2S2O8 as the oxidant (Scheme 25).48 This transformation involves an alkyl radical generated which is then added to the alkene, followed by its coupling with the phosphonyl radical.
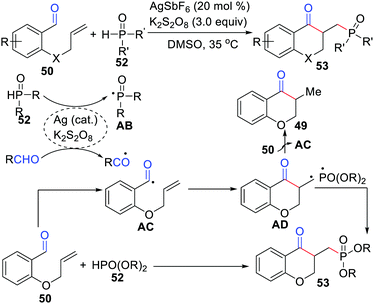 |
| | Scheme 25 Synthesis of phosphonate chroman-4-ones employed AgSbF6 as the catalyst. | |
3.6 Cyclization of 1,n-enynes
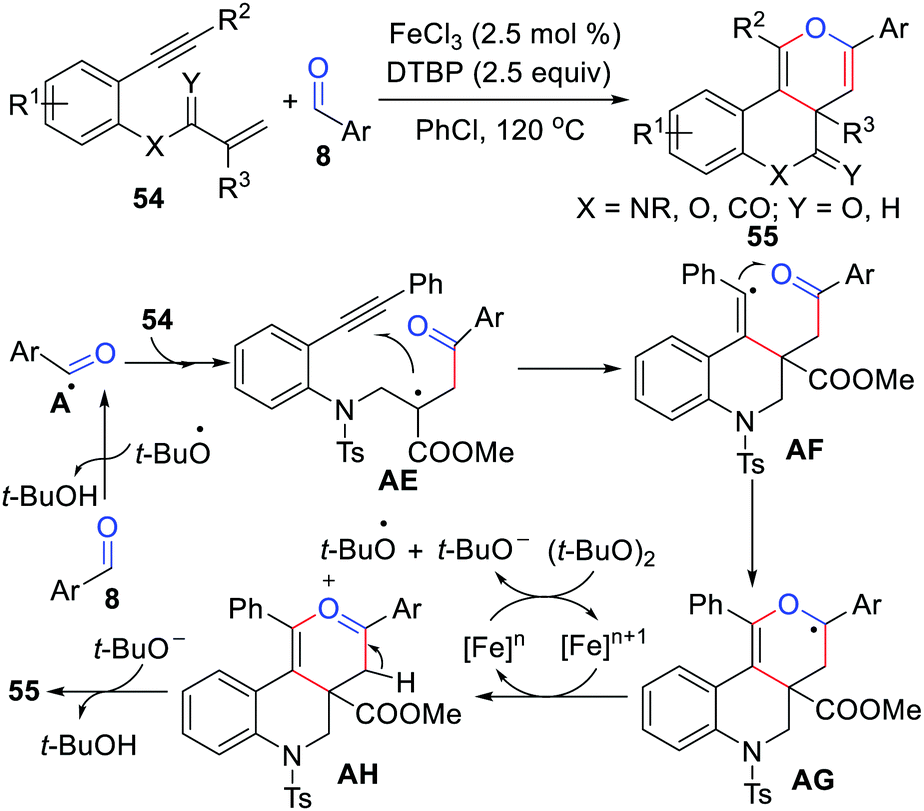
The cyclization of 1,n-enyne has been recognized as an indispensable strategy for the construction of highly intricate cyclic systems and drug molecules with elaborate ring frameworks.49 In 2016, Li's group disclosed the first example of iron-catalyzed radical [2+2+2] cyclization of 1,7-enynes with aldehydes using DTBP as the oxidant in chlorobenzene (Scheme 26).50 This reaction has a wide range tolerance of various functional groups and can synthesize fused [6.6.6] pyran derivatives in moderate yields. As shown in Scheme 26, this acyl radical-mediated method has an attractive dual role, which triggers and terminates the tandem cyclization in a one-pot procedure. Notably, Li's group also reported an iron-catalyzed [2+2+2] annulation cyclization reaction of 1,n-enynes with aldehydes in 2020.51
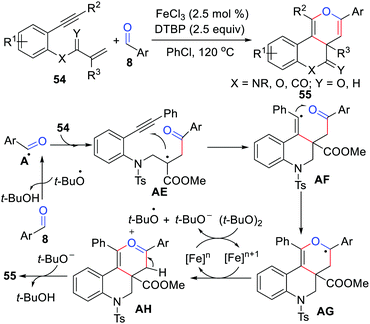 |
| | Scheme 26 Iron-catalyzed radical [2+2+2] cyclization of 1,7-enynes with aldehydes. | |
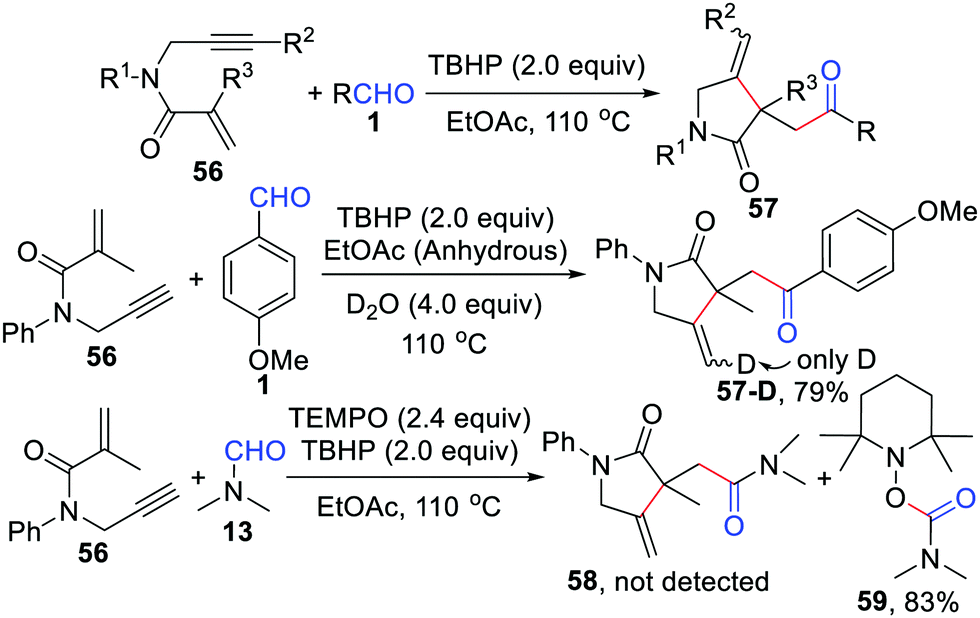
Soon after, our group also developed a regioselective radical cyclization of 1,6-enynes with aldehydes by a TBHP-mediated, one-pot process under a metal- and additive-free system (Scheme 27).52 Different from Li's iron-catalyzed strategy,50 the aldehydic radical just triggers this radical cyclization and the whole reaction is terminated by hydrogen abstraction from the solvent. One of the hydrogen atoms at the end of the olefin in the final product comes from water, which was confirmed by the isotope labeling experiment. Moreover, the acyl radical was also trapped by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) in this reaction.
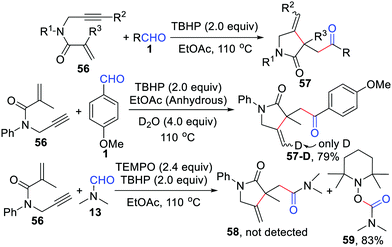 |
| | Scheme 27 Regioselective radical cyclization of 1,6-enynes with aldehydes. | |
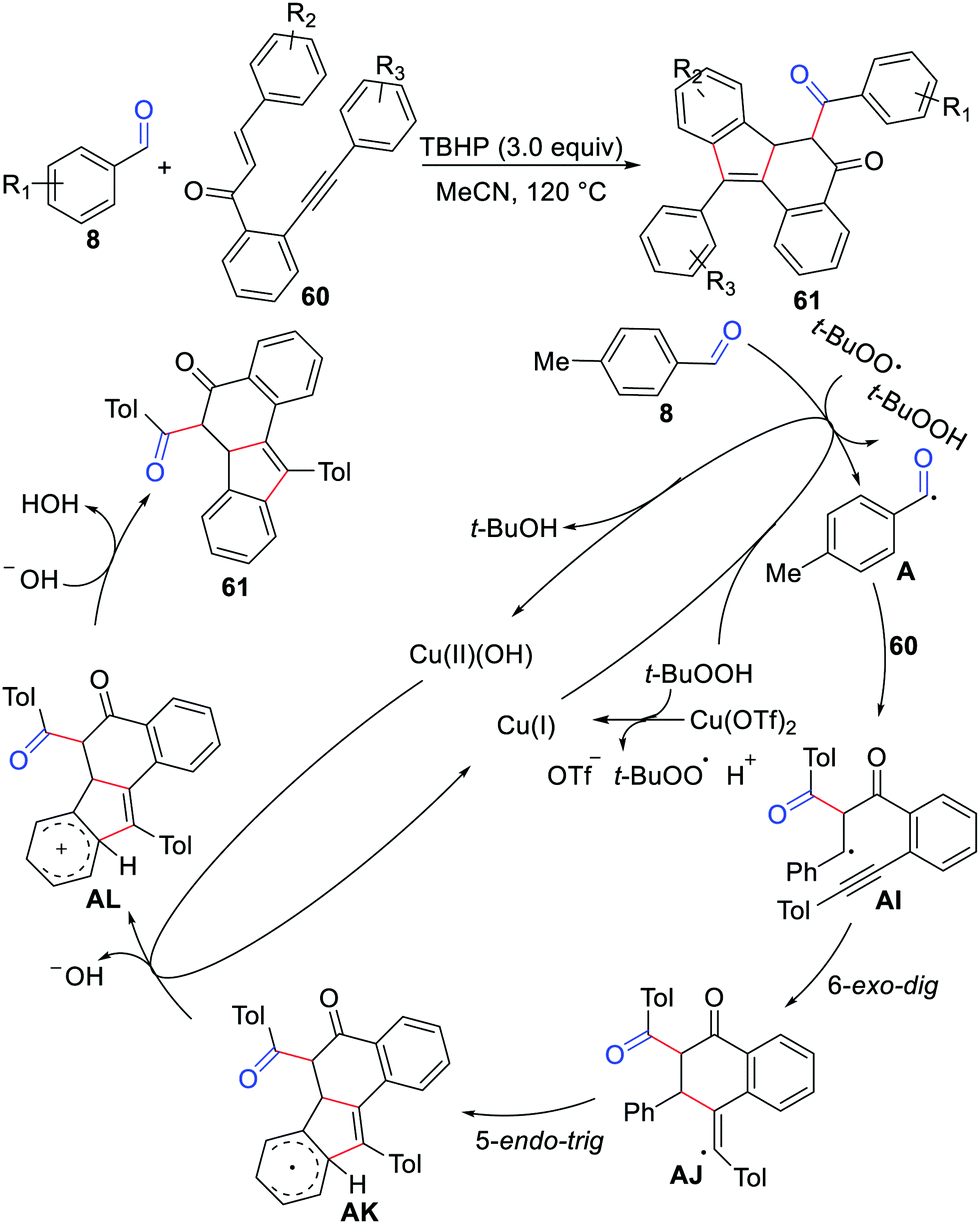
In 2020, Zhao, Wu, and co-workers described a novel bicyclization of 1,6-enynes with aldehydes for the synthesis of the 5H-benzo[a]fluoren-5-one skeleton in the presence of Cu(OTf)2 and TBHP (Scheme 28).53 The transformation constructed three C–C bonds and two carbon rings in one step with wide substrate applicability and regioselectivity. The reaction process involves 6-exo-dig and 5-endo-trig cyclization as revealed in Scheme 28.
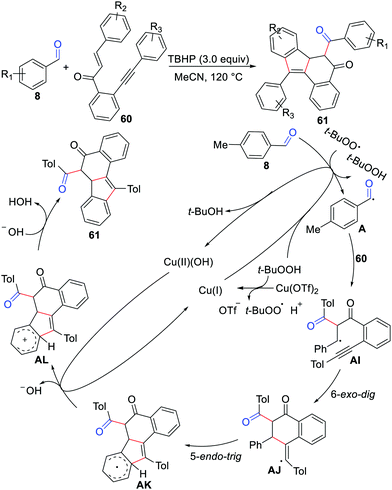 |
| | Scheme 28 Bicyclization of 1,6-enynes with aldehydes for the synthesis of the 5H-benzo[a]fluoren-5-ones. | |
3.7 Dual difunctionalization of two different alkenes
In 2019, the Yang group showed an elegant Fe-catalyzed radical dual difunctionalization of two different alkenes with aldehydes for the construction of β,δ-functionalized ketones via a one-pot procedure (Scheme 29).54 The acyl radicals produced from the aldehyde allowed for the formation of C(sp2)–C(sp3), C(sp3)–C(sp3), and C(sp3)–O bonds through dual radical insertions and radical coupling. It is worth noting that TBHP plays a triple role of radical initiator, terminal oxidant, and radical coupling partner in this reaction.
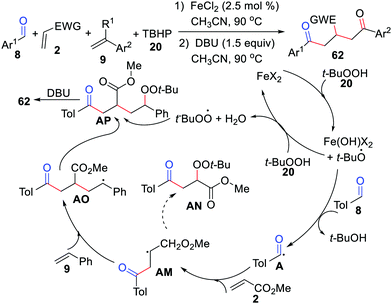 |
| | Scheme 29 Fe-Catalyzed radical dual difunctionalization of two different alkenes with aldehydes. | |
4 Conclusions
In recent years, acyl radical enabled reactions between aldehydes and alkenes have been well established and exponentially increased. The progress summarized in this review highlights the hydroacylation of simple alkenes and difunctionalization of alkenes that have been developed in organic synthesis. In these processes, the development and discovery of novel catalytic oxidation systems and reasonable design of substrate types play an important role. As shown in the above-mentioned examples, this method is quite valuable for the synthesis of diverse functionalized molecules.
However, many opportunities and challenges remain to be explored. Compared with aryl aldehyde C(sp2)–H bond cleavage, alkyl aldehyde C(sp2)–H bond cleavages under mild conditions remain relatively rare. Moreover, exploring diastereoselective radical acylation of alkenes is still a challenging target. From the perspective of synthetic applications, the application of these acyl radical enabled reactions in late-stage modifications of bioactive molecules is also a highly attractive research area. We believe more impressive methods for acyl radical enabled reactions with alkenes will be envisaged in the near future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the Natural Science Foundation of Hunan Province, China (No. 2020JJ5448), and the Scientific Research Fund of Hunan Provincial Education Department (No. 19A389) for financial support.
Notes and references
- For selected reviews, see:
(a) Y. Q. Li, D. Wu, H.-G. Cheng and G. Y. Yin, Angew. Chem., Int. Ed., 2020, 59, 7990–8003 CrossRef CAS PubMed;
(b) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. B. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed;
(c) J.-S. Zhang, L. Liu, T. Q. Chen and L.-B. Han, Chem. – Asian J., 2018, 13, 2277–2291 CrossRef CAS PubMed;
(d) J. H. Chen and Z. Lu, Org. Chem. Front., 2018, 5, 260–272 RSC;
(e) H.-Y. Tu, S. Q. Zhu, F.-L. Qing and L. L. Chu, Synthesis, 2020, 1346–1356 CAS;
(f) R. Calmanti, M. Selva and A. Perosa, Green Chem., 2021, 23, 1921–1941 RSC;
(g) A. Fingerhut, O. V. Serdyuk and S. B. Tsogoeva, Green Chem., 2015, 17, 2042–2058 RSC.
- For selected reviews, see:
(a) G. Y. S. Qiu, L. F. Lai, J. Cheng and J. Wu, Chem. Commun., 2018, 54, 10405–10414 RSC;
(b) Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC;
(c) H. Jiang and A. Studer, Chem. Soc. Rev., 2020, 49, 1790–1811 RSC;
(d) X.-W. Lan, N.-X. Wang and Y. L. Xing, Eur. J. Org. Chem., 2017, 5821–5851 CrossRef CAS;
(e) J. Lin, R.-J. Song, M. Hu and J.-H. Li, Chem. Rec., 2019, 19, 440–451 CrossRef CAS PubMed.
- For selected reviews, see:
(a) L. H. Ruan, C. X. Chen, X. X. Zhang and J. Sun, Chin. J. Org. Chem., 2018, 38, 3155–3164 CrossRef CAS;
(b) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2070 CrossRef CAS PubMed;
(c) A. Banerjee, Z. Lei and M.-Y. Ngai, Synthesis, 2019, 303–333 CAS;
(d) C. Raviola, S. Protti, D. Ravelli and M. Fagnoni, Green Chem., 2019, 21, 748–764 RSC.
- For a selected paper, see: I. Ryu, Chem. Soc. Rev., 2001, 30, 16–25 RSC.
- For selected papers, see:
(a) D. L. Boger and R. J. Mathvink, J. Org. Chem., 1992, 57, 1429–1443 CrossRef CAS;
(b) D. L. Boger and R. J. Mathvink, J. Org. Chem., 1989, 54, 1777–1779 CrossRef CAS.
- For selected papers, see:
(a) S. Bath, N. M. Laso, H. Lopez-Ruiz, B. Quiclet-Sire and S. Z. Zard, Chem. Commun., 2003, 204–205 RSC;
(b) Z. J. Liu, J. Zhang, S. L. Chen, E. Shi, Y. Xu and X. B. Wan, Angew. Chem., Int. Ed., 2012, 51, 3231–3235 CrossRef CAS PubMed;
(c) Y. J. Liu, Y. Y. Li, Y. Qi and J. Wan, Synthesis, 2010, 4188–4192 CAS;
(d) D. Crich, C. Chen, J.-T. Hwang, H. W. Yuan, A. Papadatos and R. I. Walter, J. Am. Chem. Soc., 1994, 116, 8937–8951 CrossRef CAS;
(e) C. S. Colley, D. C. Grills, N. A. Besley, S. Jockusch, P. Matousek, A. W. Parker, M. Towrie, N. J. Turro, P. M. W. Gill and M. W. George, J. Am. Chem. Soc., 2002, 124, 14952–14958 CrossRef CAS PubMed;
(f) G. W. Sluggett, C. Turro, M. W. George, I. V. Koptyug and N. J. Turro, J. Am. Chem. Soc., 1995, 117, 5148–5153 CrossRef CAS;
(g) Y. Yagci, S. P. Pappas and W. Schnabel, Z. Naturforsch., A: Phys. Sci., 1987, 42, 1425–1427 CrossRef CAS;
(h) C. E. Brown, A. G. Neville, D. M. Rayner, K. U. Ingold and J. Lusztyk, Aust. J. Chem., 1995, 48, 363–379 CrossRef CAS;
(i) A. G. Neville, C. E. Brown, D. M. Rayner, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1991, 113, 1869–1870 CrossRef CAS;
(j) M. L. Zhang, J. Xie and C. J. Zhu, Nat. Commun., 2018, 9, 3517–3527 CrossRef PubMed;
(k) R. Ruzi, K. Liu, C. J. Zhu and J. Xie, Nat. Commun., 2020, 11, 3312–3320 CrossRef CAS PubMed;
(l) R. Ruzi, J. Y. Ma, X.-A. Yuan, W. L. Wang, S. S. Wang, M. L. Zhang, J. Dai, J. Xie and C. J. Zhu, Chem. – Eur. J., 2019, 25, 12724–12729 CrossRef CAS PubMed;
(m) M. L. Zhang, X.-A. Yuan, C. J. Zhu and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 312–316 CrossRef CAS PubMed.
- For selected papers, see:
(a) L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2016, 14, 7380–7391 RSC;
(b) X.-F. Wu, Chem. – Eur. J., 2015, 21, 12252–12265 CrossRef CAS PubMed.
- For a selected paper, see: S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC.
- S. Esposti, D. Dondi, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 2531–2534 CrossRef CAS PubMed.
-
(a) S. A. Moteki, A. Usui, S. Selvakumar, T. X. Zhang and K. Maruoka, Angew. Chem., Int. Ed., 2014, 53, 11060–11064 CrossRef CAS PubMed;
(b) E. Voutyritsa and C. G. Kokotos, Angew. Chem., Int. Ed., 2020, 59, 1735–1741 CrossRef CAS PubMed;
(c) M. D. Vu, M. Das and X.-W. Liu, Chem. – Eur. J., 2017, 23, 15899–15902 CrossRef CAS PubMed;
(d) G. N. Papadopoulos, E. Voutyritsa, N. Kaplaneris and C. G. Kokotos, Chem. – Eur. J., 2018, 24, 1726–1731 CrossRef CAS PubMed;
(e) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, ChemSusChem, 2019, 12, 4194–4201 CrossRef CAS PubMed;
(f) J. L. Jiang, R. Ramozzi, S. Moteki, A. Usui, K. Maruoka and K. Morokuma, J. Org. Chem., 2015, 80, 9264–9271 CrossRef CAS PubMed;
(g) D. Ravelli, M. Zema, M. Mella, M. Fagnoni and A. Albinia, Org. Biomol. Chem., 2010, 8, 4158–4164 RSC;
(h) X. M. Wang, Y. M. Chen, H. J. Song, Y. X. Liu and Q. M. Wang, Org. Lett., 2021, 23, 2199–2204 CrossRef CAS PubMed;
(i) H. W. Liu, F. Xue, M. Wang, X. X. Tang and J. Wu, Synlett, 2021, 406–410 CAS;
(j) P. Fan, Y. Lan, C. Zhang and C. Wang, J. Am. Chem. Soc., 2020, 142, 2180–2186 CrossRef CAS PubMed;
(k) S. Paul and J. Guin, Chem. – Eur. J., 2021, 27, 4412–4419 CrossRef CAS PubMed;
(l) X.-Z. Fan, J.-W. Rong, H.-L. Wu, Q. Zhou, H.-P. Deng, J. D. Tan, C.-W. Xue, L.-Z. Wu, H.-R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed;
(m) P. Fan, C. Zhang, Y. Lan, Z. Y. Lin, L. C. Zhang and C. Wang, Chem. Commun., 2019, 55, 12691–12694 RSC.
- S. Selvakumar, R. Sakamoto and K. Maruoka, Chem. – Eur. J., 2016, 22, 6552–6555 CrossRef CAS PubMed.
- S. Tsujimoto, T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 2001, 2352–2353 RSC.
- For selected reviews, see:
(a) M. G. J. Doyle and R. J. Lundgren, Chem. Commun., 2021, 57, 2724–2731 RSC;
(b) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed;
(c) G. P. Mcglacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447–2464 RSC;
(d) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540–548 RSC;
(e) C. Liu, H. Zhang, W. Shi and A. W. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed;
(f) A. K. Bagdi, M. Rahman, D. Bhattacherjee, G. V. Zyryanov, S. Ghosh, O. N. Chupakhin and A. Hajra, Green Chem., 2020, 22, 6632–6681 RSC.
- J. Wang, C. Liu, J. W. Yuan and A. W. Lei, Angew. Chem., Int. Ed., 2013, 52, 2256–2259 CrossRef CAS PubMed.
- K. Zhao, X.-C. Zhang, J.-Y. Tao, X.-D. Wu, J.-X. Wu, W.-M. Li, T.-H. Zhu and T.-P. Loh, Green Chem., 2020, 22, 5497–5503 RSC.
- J.-W. Yuan, Q.-Y. Yin, L.-R. Yang, W.-P. Mai, P. Mao, Y.-M. Xiao and L.-B. Qu, RSC Adv., 2015, 5, 88258–88265 RSC.
- M. Adib, R. Pashazadeh, S. Rajai-Daryasarei, R. Kabiri and M. Jahani, RSC Adv., 2016, 6, 110656 RSC.
- X.-H. Yang, W.-T. Wei, H.-B. Li, R.-J. Song and J.-H. Li, Chem. Commun., 2014, 50, 12867–12869 RSC.
- S. Tripathi, R. Kapoor and L. D. S. Yadav, Adv. Synth. Catal., 2018, 360, 1407–1413 CrossRef CAS.
- For selected papers, see:
(a) W. Adam, C. R. Saha-Möller and P. A. Ganeshpure, Chem. Rev., 2001, 101, 3499–3548 CrossRef CAS PubMed;
(b) Q. H. Xia, H. Q. Ge, C. P. Ye, Z. M. Liu and K. X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS PubMed;
(c) M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed;
(d) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- Q. P. Ke, B. Y. Zhang, B. L. Hu, Y. X. Jin and G. Z. Lu, Chem. Commun., 2015, 51, 1012–1015 RSC.
- W.-T. Wei, X.-H. Yang, H.-B. Li and J.-H. Li, Adv. Synth. Catal., 2015, 357, 59–63 CrossRef CAS.
-
(a) G. F. P. de Souza, J. A. Bonacin and A. G. Salles, J. Org. Chem., 2018, 83, 8331–8340 CrossRef CAS PubMed;
(b) J. Li and D. Z. Wang, Org. Lett., 2015, 17, 5260–5263 CrossRef CAS PubMed;
(c) V. Ashokkumar and A. Siva, Org. Biomol. Chem., 2017, 15, 2551–2561 RSC.
- For selected reviews, see:
(a) Z. Y. Wu, M. Hu, J. X. Li, W. Q. Wu and H. F. Jiang, Org. Biomol. Chem., 2021, 19, 3036–3054 RSC;
(b) Y.-C. Wu, Y.-T. Xiao, Y.-Z. Yang, R.-J. Song and J.-H. Li, ChemCatChem, 2020, 12, 5312–5329 CrossRef CAS;
(c) C. J. R. Bataille and T. J. Donohoe, Chem. Soc. Rev., 2011, 40, 114–128 RSC;
(d) E. M. Beccalli, G. Broggini, M. Martinelli and S. Sottocornola, Chem. Rev., 2007, 107, 5318–5365 CrossRef CAS PubMed.
- W. P. Liu, Y. M. Li, K. S. Liu and Z. P. Li, J. Am. Chem. Soc., 2011, 133, 10756–10759 CrossRef CAS PubMed.
- K. S. Liu, Y. M. Li, X. J. Zheng, W. P. Liu and Z. P. Li, Tetrahedron, 2012, 68, 10333–10337 CrossRef CAS.
- L. L. Zhao, Y. Wang, Z. L. Ma and Y. H. Wang, Inorg. Chem., 2017, 56, 8166–8174 CrossRef CAS PubMed.
- W.-C. Yang, S.-S. Weng, A. Ramasamy, G. Rajeshwaren, Y.-Y. Liao and C.-T. Chen, Org. Biomol. Chem., 2015, 13, 2385–2392 RSC.
- L. Ge, Y. J. Li and H. L. Bao, Org. Lett., 2019, 21, 256–260 CrossRef CAS PubMed.
- L. Zhang, S. Y. Liu, Z. G. Zhao, H. M. Su, J. C. Hao and Y. Wang, Chem. Sci., 2018, 9, 6085–6090 RSC.
- Y. H. Jiao, M.-F. Chiou, Y. J. Li and H. L. Bao, ACS Catal., 2019, 9, 5191–5197 CrossRef CAS.
- M.-B. Zhou, R.-J. Song, X.-H. Ouyang, Y. Liu, W.-T. Wei, G.-B. Deng and J.-H. Li, Chem. Sci., 2013, 4, 2690–2694 RSC.
-
(a) F. Jia, K. S. Liu, H. Xi, S. L. Lu and Z. P. Li, Tetrahedron Lett., 2013, 54, 6337–6340 CrossRef CAS;
(b) B. Niu, L. Xu, P. Xie, M. Wang, W. N. Zhao, C. U. P. Jr. and A. H. Zhou, ACS Comb. Sci., 2014, 16, 454–458 CrossRef CAS PubMed;
(c) W. W. Gong, L. Xu, T. Ji, P. Xie, X. Y. Qi, C. U. P. Jr. and A. H. Zhou, RSC Adv., 2014, 4, 6854–6857 RSC;
(d) R. Sakamoto, N. Hirama and K. Maruoka, Org. Biomol. Chem., 2018, 16, 5412–5415 RSC;
(e) R. Boora, G. R. Kumar and B. V. S. Reddy, Org. Biomol. Chem., 2019, 17, 9627–9630 RSC;
(f) P. Biswas, S. Mandal and J. Guin, Org. Lett., 2020, 22, 4294–4299 CrossRef CAS PubMed.
- W. N. Zhao, P. Xie, M. Zhang, B. Niu, Z. G. Bian, C. P. Jr. and A. H. Zhou, Org. Biomol. Chem., 2014, 12, 7690–7693 RSC.
- W.-P. Mai, J.-T. Wang, Y.-M. Xiao, P. Mao and K. Lu, Tetrahedron, 2015, 71, 8041–8051 CrossRef CAS.
- Z.-Q. Xu, C. Wang, L. Li, L. L. Duan and Y.-M. Li, J. Org. Chem., 2018, 83, 9718–9728 CrossRef CAS PubMed.
- L. Y. Lv, L. Y. Qi, Q. X. Guo, B. J. Shen and Z. P. Li, J. Org. Chem., 2015, 80, 12562–12571 CrossRef CAS PubMed.
- Y. N. Li, F. Y. Ying, T. F. Fu, R. H. Yang, Y. Dong, L. Q. Lin, Y. H. Han, D. Q. Liang and X. H. Long, Org. Biomol. Chem., 2020, 18, 5660–5665 RSC.
- L. Y. Lv, H. Xi, X. H. Bai and Z. P. Li, Org. Lett., 2015, 17, 4324–4327 CrossRef CAS PubMed.
- L. Y. Lv and Z. P. Li, Chin. J. Chem., 2017, 35, 303–306 CrossRef CAS.
- L. Y. Lv, S. L. Lu, Q. X. Guo, B. J. Shen and Z. P. Li, J. Org. Chem., 2015, 80, 698–704 CrossRef CAS PubMed.
- Y. Liu, Q.-L. Wang, C.-S. Zhou, B.-Q. Xiong, P.-L. Zhang, C.-A. Yang and K.-W. Tang, J. Org. Chem., 2018, 83, 4657–4664 CrossRef CAS PubMed.
- T. Tian, X. Wang, L. Y. Lv and Z. P. Li, Chem. Commun., 2020, 56, 14637–14640 RSC.
- C. D. Pan, Q. T. Ni, Y. Fu and J.-T. Yu, J. Org. Chem., 2017, 82, 7683–7688 CrossRef CAS PubMed.
- Y.-M. Xiao, Y. Liu, W.-P. Mai, P. Mao, J.-W. Yuan and L.-R. Yang, ChemistrySelect, 2019, 4, 1939–1942 CrossRef CAS.
- H.-S. Jhuang, D. M. Reddy, T.-H. Chen and C.-F. Lee, Asian J. Org. Chem., 2016, 5, 1452–1456 CrossRef CAS.
- S. Jung, J. Kim and S. Hong, Adv. Synth. Catal., 2017, 359, 3945–3949 CrossRef CAS.
- J. J. Zhao, P. Li, X. J. Li, C. G. Xia and F. W. Li, Chem. Commun., 2016, 52, 3661–3664 RSC.
- For selected reviews and a paper, see:
(a) J. Xuan and A. Studer, Chem. Soc. Rev., 2017, 46, 4329–4346 RSC;
(b) C.-H. Xu, Y. Li, J.-H. Li, J.-N. Xiang and W. Deng, Sci. China: Chem., 2019, 62, 1463–1475 CrossRef CAS;
(c) Y. Li, G.-A. Pan, M.-J. Luo and J.-H. Li, Chem. Commun., 2020, 56, 6907–6924 RSC;
(d) W.-T. Wei, Q. Li, M.-Z. Zhang and W.-M. He, Chin. J. Catal., 2021, 42, 731–742 CrossRef CAS;
(e) X.-X. Meng, Q.-Q. Kang, J.-Y. Zhang, Q. Li, W.-T. Wei and W.-M. He, Green Chem., 2020, 22, 1388–1392 RSC.
- L. Y. Lv and Z. P. Li, Org. Lett., 2016, 18, 2264–2267 CrossRef CAS PubMed.
- T. Tian, X. Wang, L. Y. Lv and Z. P. Li, Eur. J. Org. Chem., 2020, 4425–4428 CrossRef CAS.
- X.-D. Xu, T.-T. Cao, Y.-N. Meng, G. D. Zhou, Z. Y. Guo, Q. Li and W.-T. Wei, ACS Sustainable Chem. Eng., 2019, 7, 13491–13496 CrossRef CAS.
- Y. Zhou, P. Zhao, X.-X. Yu, X. Geng, C. Wang, C. Huang, H. Yang, Z.-Y. Zhao and A.-X. Wu, Org. Lett., 2020, 22, 8359–8364 CrossRef CAS PubMed.
- C.-S. Wu, R.-X. Liu, D.-Y. Ma, C.-P. Luo and L. Yang, Org. Lett., 2019, 21, 6117–6121 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c and
Wen-Ting
Wei
*c and
Wen-Ting
Wei