
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDe novo designed coiled coils as scaffolds for lanthanides, including novel imaging agents with a twist
Alexandra M.
Webster
 * and
Anna F. A.
Peacock
*
* and
Anna F. A.
Peacock
*
School of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: alexamwebster@gmail.com; a.f.a.peacock@bham.ac.uk
First published on 15th June 2021
Abstract
For much of their history, lanthanides were thought to be biologically inert. However, the last decade has seen the discovery and development of the field of native lanthanide biochemistry. Lanthanides exhibit a variety of interesting photophysical properties from which many useful applications derive. The development of effective functional lanthanide complexes requires control of their coordination sphere; something proteins manage very effectively through their 3D metal-binding sites. α-Helical coiled coil peptides are miniature scaffolds which can be designed de novo and can retain the favourable properties of larger proteins within a much simplified system. Metal binding sites, including those which bind lanthanides can be engineered into the coiled coil sequence. This review will highlight the opportunities presented by the use of coiled coil peptides as scaffolds for lanthanide binding and the potential to control the coordination environment by simple modifications to peptide sequence. Designed lanthanide coiled coils offer opportunities to gain greater insight into native lanthanide biochemistry as well as to develop new functional complexes, including imaging agents.
Introduction
Once described as ‘boring’1 the spectroscopic and photophysical properties of the lanthanides means they have a number of useful applications, including, amongst others, as luminescent probes,2 as paramagnetic shift agents for NMR spectroscopy,3 as probes for structure determination via X-ray crystallography4 and as MRI contrast agents.5 The recent discovery of native lanthanide biochemistry further contradicts this unfavourable historical view.6,7Metal-binding proteins are ubiquitous in nature; 30–40% of proteins are thought to require a metal to carry out their function.8 Proteins act as sophisticated ligands which are able to precisely control metal coordination chemistry, thereby tuning protein function.9–13 Thus harnessing the power of proteins as ligands for metal ions is an attractive prospect and an active area of research.14–19 One strategy is the design of structured peptides de novo, whereby miniaturized scaffolds with metal-binding sites that replicate the environment found within more complex proteins are engineered. Although various peptide folds have been successfully designed de novo, including β-hairpins20 and γ-turns,21 the majority of work has focussed on the design of α-helical assemblies, including coiled coils, which can be more predictably designed.
Herein, we will provide a brief overview of the photophysical, spectroscopic and biochemical properties of the lanthanides before discussing the development of lanthanide-binding coiled coils and, importantly, how this new class of ligands can control the lanthanide coordination chemistry and, in turn, the physical properties.
Luminescence
Lanthanide luminescence arises from f–f transitions which are formally Laporte forbidden.22 As a result of weak ligand interactions, the emission bands of lanthanide complexes are characteristically narrow. Furthermore, due to the parity (and often spin) forbidden electronic transitions, lanthanide luminescence is long lived with μs to ms timescales. Moreover, there is no significant quenching from oxygen, meaning that even in solution, at room temperature and in the presence of oxygen, for lanthanides such as Eu3+ and Tb3+ radiative lifetimes are long, facilitating time-resolved emission experiments. However, lanthanide luminescence is weak with low extinction coefficients (ε is typically ≪10 mol−1 dm3 cm−1)23,24 and therefore lanthanide probes usually incorporate sensitising chromophores to enhance emission via the antenna effect.22,25,26Whilst the lanthanide excited states are not significantly quenched by oxygen, they are very efficiently quenched by water, specifically the O–H stretching vibration.24,27 This means that in order to optimise lanthanide luminescence in aqueous solutions, metal-bound water molecules must be avoided (or minimised). This is made more difficult by the high affinity of the Ln3+ ions for water; the magnitude of their ΔHhydr values are high (ca. −3300 to −3700 kJ mol−1).28 As a result, lanthanide complexes with low denticity ligands rapidly dissociate in aqueous solutions to form aqua ions [Ln(H2O)n]3+, where n = 9 for La3+ to Gd3+, and then decreases to 8 moving along the series to Lu3+.29 These aqua ions are also prone to hydrolysis to their insoluble hydroxides, and a slightly acidic pH is needed to inhibit this process.30 In order to optimise lanthanide luminescence under aqueous conditions, complexes must possess both high kinetic and thermodynamic stability. To achieve this, polydentate ligands containing hard oxygen and nitrogen donors are used to improve thermodynamic stability via the chelate effect, and rigid ligands, in particular macrocyclic ligands, improve kinetic stability.
NMR applications of lanthanides
With the exception of La3+ and Lu3+ ([Xe]4f0 and [Xe]4f14 respectively), all Ln3+ ions are paramagnetic. Usually paramagnetic species are excluded from samples for NMR spectroscopy as they are associated with large chemical shift ranges and significant broadening of signals.31 Paramagnetic lanthanides also cause shifts in NMR resonances but it is to a more manageable extent. With the exception of Gd3+, line broadening is not as severe; in particular, for Eu3+ and Yb3+ resonances remain sharp.3,32 Various lanthanide shift reagents are commercially available and addition to an NMR sample can simplify complex spectra by resolving the peaks. Additionally, complexation of lanthanides to proteins, either via an existing metal binding site or via use of a prosthetic lanthanide coordinating group, is used to provide valuable structural insights by way of long range (∼40 Å) distance and angular information.4,33–36A related application is in MRI (magnetic resonance imaging). MRI is an imaging technique based on NMR which is widely used in clinical medicine to provide detailed information about different tissues. Contrast agents are used to enhance images generated by MRI by shortening the relaxation times of water protons. Contrast agents aim to alleviate two problems associated with MRI techniques. First, by increasing relaxivity and thereby decreasing the relaxation time of water protons, scans may be acquired more quickly. Second, accumulation of the contrast agent in particular tissues enables differentiation between tissues that would otherwise be difficult to distinguish from one another. The development of molecular imaging has led to the generation of ‘targeted’ and ‘smart’ contrast agents.37 Targeted contrast agents are designed to accumulate inside specific types of tissue whereas smart contrast agents respond to biological processes. For example, a targeted contrast agent may be conjugated to a ligand which binds to receptors found on the surface of particular cell types and thus reduce relaxation times of nearby protons, while a smart contrast agent will have no effect on proton relaxation times until interaction with species generated by certain biological processes.
Ln3+ complexes, especially Gd3+ may be used as contrast agents. Gd3+ has seven unpaired electrons, the most of any known elemental species, and may decrease T1 relaxation times by a factor of up to 106, making it the most commonly used species in MRI contrast agents.38 However, consideration of toxicity of free Gd3+ is necessary. Ln3+ ions are a similar size to Ca2+, and like Ca2+, prefer hard oxygen ligands. This means that lanthanides can interfere with calcium-dependent processes such as muscular contraction and neurotransmission. Free Ln3+ ions are also excreted slowly and accumulate in liver, spleen, kidney and bone tissue.39 Hence, gadolinium contrast agents must be stable to dissociation under physiological conditions, and as such, multidentate, macrocyclic ligands are prevalent in current clinically used gadolinium contrast agents.38–40
Whilst the binding affinity of Gd3+ for small molecule macrocyclic ligands is high (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K ∼ 23), relaxivity is suboptimal. Only one water molecule tends to be directly coordinated to Gd3+ and the rate of water exchange is slower than the Larmor frequency for machines used in the clinic. Additionally, the rapid molecular tumbling of small molecules in solution has a detrimental effect on relaxivity. The result is that in order to be effective contrast agents, gram quantities must be administered,41 and as such there is a desire for the development of more effective contrast agents. An increase in the hydration state of Gd3+, optimising water exchange rate and molecular tumbling rate are all approaches that can be adopted to improve relaxivity. Peptide-based contrast agents where coordination environment can be carefully controlled could therefore prove to be promising. Moreover, designs featuring peptides have shown promise as targeted contrast agents.42
K ∼ 23), relaxivity is suboptimal. Only one water molecule tends to be directly coordinated to Gd3+ and the rate of water exchange is slower than the Larmor frequency for machines used in the clinic. Additionally, the rapid molecular tumbling of small molecules in solution has a detrimental effect on relaxivity. The result is that in order to be effective contrast agents, gram quantities must be administered,41 and as such there is a desire for the development of more effective contrast agents. An increase in the hydration state of Gd3+, optimising water exchange rate and molecular tumbling rate are all approaches that can be adopted to improve relaxivity. Peptide-based contrast agents where coordination environment can be carefully controlled could therefore prove to be promising. Moreover, designs featuring peptides have shown promise as targeted contrast agents.42
The native biochemistry of lanthanides
Until recently, lanthanides were not considered biologically relevant metals. Whilst early lanthanides are fairly abundant in the earth's crust, and whilst the similarities in the size and bonding preference with known biologically active metals such as calcium had been well documented, the low aqueous solubility of lanthanides in the environment was thought to make their use in native biochemistry unlikely.43 However, in 2011, it was found that addition of La3+ to the culture medium of bacteria Methylobacterium radiotolerans caused the xoxF gene (whose function was hitherto unknown) to express a methanol dehydrogenase-like enzyme.44 Since then, many bacteria containing the xoxF gene have been identified, including lanthanide dependent extremophiles45 as well as methylotrophic bacteria abundant in marine environments,46 in soils47 and in the phyllospheres of plants.48 The emergence of the role of lanthanides in native biochemistry presents the potential for engineering new oxidation catalysts and new systems for detecting and extracting lanthanides from the environment. The field of native lanthanide biochemistry is new but quickly developing, and progress in the field has been extensively reviewed several times recently.6,7,49,50Given that lanthanides are now recognised as biologically relevant metal ions, there is a need to expand our understanding of lanthanide biochemistry, and given the opportunities afforded by their chemical properties, there is an opportunity to develop new lanthanide-based biological agents. Harnessing control of the local lanthanide environment is paramount if either of these goals are to be achieved, and several strategies have been attempted as a means to do this in peptide-based systems.
Inspired by naturally occurring calcium binding loops: lanthanide binding tags
Given the similarity of trivalent lanthanides to divalent calcium – both share similar ionic radii and preferences for oxygen-based ligands – it is no surprise that researchers should look to naturally occurring calcium-binding sites when designing lanthanide binding peptides. One such example is the EF hand motif which is a structural domain found in many calcium binding proteins and consists of two α-helices linked by a short calcium binding loop (Fig. 1).51 Several studies have shown that La3+ binds to the same EF domains that bind Ca2+.52–55 Indeed, the EF hand motif provided the starting point for the development of short peptide sequences (≤20 residues) capable of high affinity lanthanide binding known as lanthanide binding tags (LBTs, Fig. 1).4,56–58 Dissociation constants for LBTs may be in the low nM range,59 and form a highly ordered chelate structure which generally excludes water from the lanthanide inner coordination sphere,60 thus minimising luminescence quenching. LBTs may be introduced at either the C- or N-termini of proteins, or encoded within existing loops and turns in a peptide sequence.61,62 In this way, lanthanides may be incorporated within peptides and proteins which lack suitable binding loops in their native sequence. This assists with characterisation of peptides and proteins. For example, LBTs have been used to solve the phase problem in X-ray crystallography63 and in NMR spectroscopy to provide long-distance and angular information.64,65 The luminescent properties of the majority of the lanthanides mean that LBTs can also be used to provide information on protein function and dynamics.66,67 Finally, a gadolinium-binding LBT was engineered to include a single water molecule within the metal inner coordination sphere and was reported to display longitudinal relaxivity (r1) values comparable to clinically used contrast agents gadopentate dimeglumine (Magnevist) and gadodiamide (Omniscan) at 11.7 T.68 This LBT could be co-expressed with a range of proteins in order to provide, in principle, a route to targeted MRI contrast agents.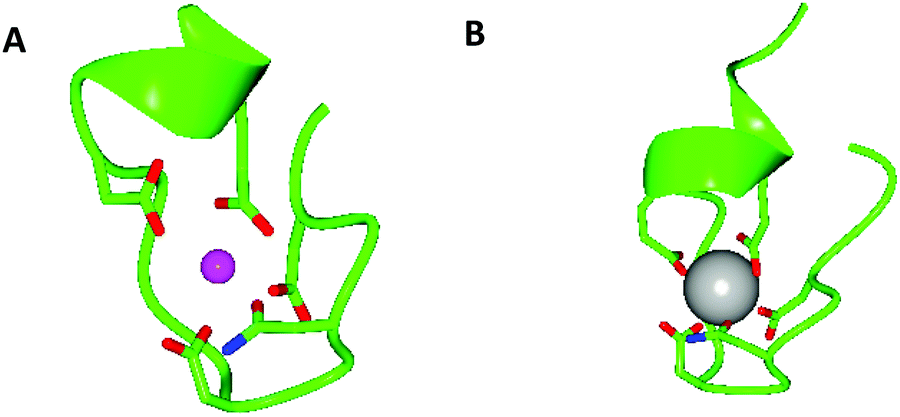 | ||
| Fig. 1 Models generated using CCP4 software,134,135 oxygen atoms are red, nitrogen atoms are blue, Ca2+ is a pink sphere and Gd3+ is a grey sphere. (A) X-ray crystal structure of the 52 kDa domain of human cardiac troponin, residues 104–117, showing coordination of the Ca2+ binding site of one of the EF hand motifs of human cardiac troponin with coordinating side chains. Coordinating backbone oxygen omitted for clarity, pdb reference 1J1E.136 (B) X-ray crystal structure of a lanthanide binding tag showing the coordination environment of bound Gd3+ and coordinating side chains, coordinating backbone oxygen omitted for clarity, pdb reference 3VDZ.60 | ||
More recently it has been found that, rather than simply being capable of binding La3+ in a laboratory environment, EF hand motifs are present in proteins that natively bind La3+. Lanmodulin is a recently discovered bacterial protein containing four EF hand motifs, three of which bind lanthanides with picomolar affinities (the fourth with micromolar affinities), and shows a hundred million fold selectivity for La3+ over Ca2+.69 In 2019, Cotruvo and co-workers designed a fluorescent sensor which used lanmodulin to link two proteins as a Förster Resonance Energy Transfer (FRET) pair. The sensor was found to bind lanthanides with picomolar dissociation constants and only ∼2 fold weaker than the native lanmodulin.70
LBTs are not the only peptide or protein-based lanthanide binding agents. Recently, a TIM barrel protein which consisted of several de novo designed domains was shown to bind lanthanides with femtomolar dissociation constants.71 Other peptide lanthanide binding strategies include the use of cyclic peptides,72,73 the modification of zinc finger proteins,74–76 phosphorylated peptides77,78 and the use of non-natural amino acids.79 However a full discussion of these is beyond the scope of this perspective unless relevant to the peptide coiled coil scaffolds discussed.
De novo designed peptide coiled coils as ligands
Nature very effectively regulates protein function by controlling metal ion primary and secondary coordination sphere effects.9 However, metalloproteins are large (average size ∼50 kDa), complex systems and as such it can be a challenge to interpret the chemical observations with a detailed understanding of the chemistry at the atomic level. The de novo design of peptides sequences which adopt defined and predictable secondary, tertiary and quaternary structures, provides a route to generating highly organised systems which possess many of the features of native proteins, whilst retaining some of the simplicity of small molecules. The most common of these miniature scaffolds are based on the α-helix, including the coiled coil. Coiled coils are supercoils of two or more α-helices wrapped around each other in a left-handed manner, based on well-established design rules. In spite of their relative simplicity, coiled coils are highly organised structures in which well defined ‘active sites’ can be engineered.80–86 In addition to the lanthanides which will be covered in this review, examples of other metals that have been incorporated into coiled coils include, but are not limited to, first row transition metals such as zinc,87 iron,88 copper,89 and heavy metals associated with toxicity such as mercury90 and cadmium,91 as well as systems incorporating multiple metal binding sites.92–95One advantage of coiled coil ligands is the capacity to control both the primary and secondary coordination spheres of bound metals by small changes in the amino acid sequence. Control of the secondary coordination sphere can be difficult to regulate in a predictable manner in small molecule systems, but can have a significant influence on a metal's physical and chemical properties. For example, it can alter the catalytic activity of metal complexes by controlling selectivity and/or reactivity.9,96–98 This can occur through preorganisation of first coordination sphere ligands,99 steric encumbrance,100 and better interaction with biological targets.101 Specific to lanthanide complexes, secondary coordination sphere effects in small molecules complexes have been shown to alter their stability,102 luminescent101 and magnetic properties.104 Thus it is clear that control of the secondary coordination sphere is highly desirable, and something which is achievable with coiled coil peptide systems.
Principles of peptide coiled coil design
The principles of coiled coil design have been reviewed in detail several times,105–107 and so only a brief overview is provided below.Peptide α-helical coiled coil design is based on a system of repeating heptads, denoted as (abcdefg)n where a–g are amino acid residues and n is the number of heptads (generally n ≥ 3 to promote folding). Generally, if the amino acids in the a and d positions are hydrophobic, this creates a hydrophobic face. Assembly of multiple α-helices into a coiled coil is driven by burying these residues, making a hydrophobic core (Fig. 2). Each heptad therefore contains two turns, but since this is not exact – there are 3.6 residues per turn – the hydrophobic face migrates around the α-helix in the opposite direction to the helix backbone leading to supercoiling.
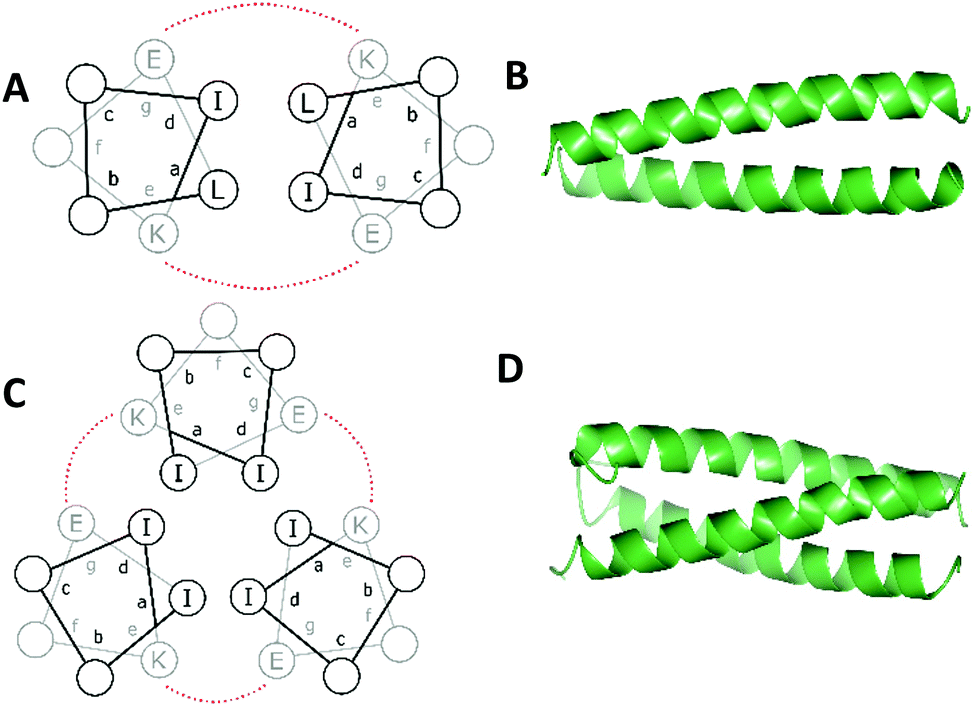 | ||
| Fig. 2 Helical wheel diagrams of a (A) two- and (C) three-stranded coiled coil. Hydrophobic residues are black and salt bridge interactions between g and e residues are shown (red). X-ray crystal structures of a de novo designed homo- (B) dimer (pdb 1ZII) and (D) trimer (pdb 1ZIJ).137 Models generated using CCP4 software.134,135 | ||
Packing of the hydrophobic a and d residues determines the number of α-helices making up the coiled coil. Generally, a = isoleucine (Ile) and d = leucine (Leu) promotes the formation of dimeric coiled coils, a = d = Ile or Leu promotes the formation of coiled coil trimers and a = Leu and d = Ile promotes the formation of tetrameric coiled coils.108 Valine (Val) or alanine (Ala) may also be used.
As illustrated for a parallel three stranded coiled coil in Fig. 2, charged residues in the g position of one α-helix interact with complementary residues in the e position of another in an i – i′ + 5, or 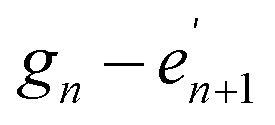 manner, forming interhelical salt bridges. Residues in the b and c positions tend to be water solubilising groups and/or helix promoters.109 Finally, the f position is least critical to coiled coil formation and as such provides a site where other functionality can be introduced. Examples have included unnatural amino acids that provide an opportunity for labelling (for example, azides and alkynes for copper catalysed azide–alkyne click reactions),110 or groups that aid coiled coil characterisation, such as 4-iodo-L-phenylalanine for X-ray crystallography.106
manner, forming interhelical salt bridges. Residues in the b and c positions tend to be water solubilising groups and/or helix promoters.109 Finally, the f position is least critical to coiled coil formation and as such provides a site where other functionality can be introduced. Examples have included unnatural amino acids that provide an opportunity for labelling (for example, azides and alkynes for copper catalysed azide–alkyne click reactions),110 or groups that aid coiled coil characterisation, such as 4-iodo-L-phenylalanine for X-ray crystallography.106
Metal-binding sites can be engineered externally or more commonly within the hydrophobic core of the coiled coil, and, through a process of peptide design, the coordination chemistry of the bound metal tuned. Given the recent recognition of the importance of understanding lanthanide biochemistry, and the established work demonstrating that engineered lanthanide sites are extremely useful spectroscopic tools, our group and others have explored lanthanide coordination to miniature de novo designed protein scaffolds, including the coiled coil.
Lanthanides at the interhelical interface
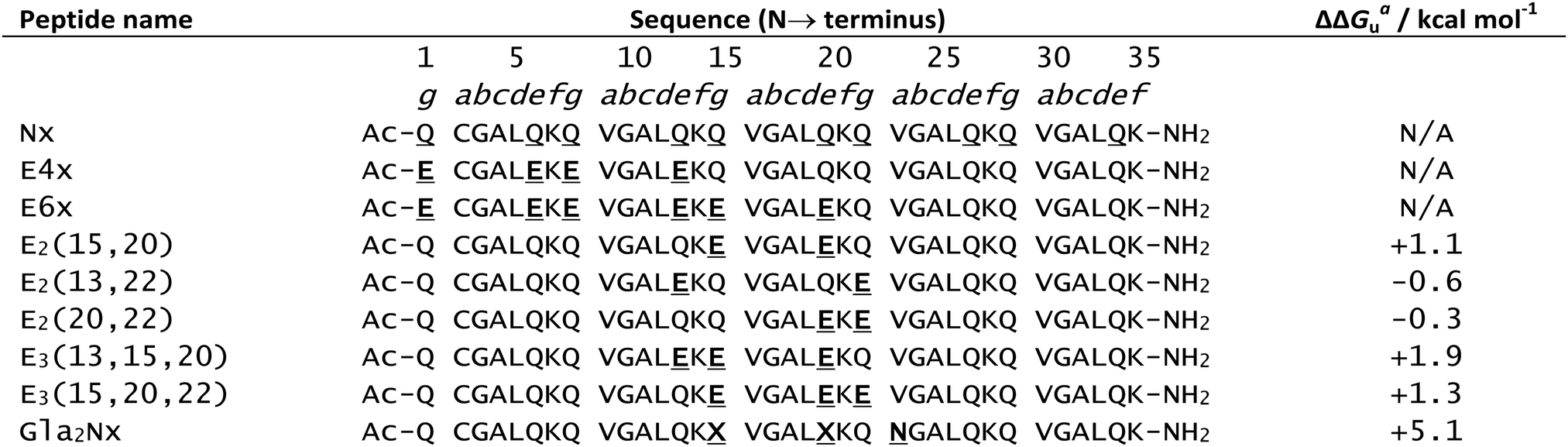
In 1996, Hodges and co-workers investigated the effects of metal binding on relieving interhelical electrostatic repulsions in coiled coils.111 Based on the sequence of Nx, a peptide known to adopt a dimeric coiled coil structure, Table 1, a series of peptides were synthesised in which there were varying number of negatively charged (at physiological pH) Glu for neutral Gln substitutions, named Enx where n = 4, 6, 8 or 10 and represents the number of Glu for Gln substitutions (Table 1). These substitutions resulted in Glu residues in the e and g positions which electrostatically repel each other, disfavouring coiled coil formation. Accordingly, increasing the number of Gln to Glu substitutions correlated with gradual loss of helical content and stability. Addition of KCl or LaCl3, restored helicity to all peptides, however the coiled coils formed on addition of LaCl3 were notably more stable than those formed on addition of KCl. This difference became more marked as the number of Glu residues increased, consistent with direct interaction of La3+ ions with Glu residues, countering the negative charge repulsion. In contrast, monovalent K+ promotes coiled coil formation via inducing a greater hydrophobic effect, but this is not strong enough to completely overcome interhelical charge repulsion.![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) represents uncommon amino acid Gla. Coiled coil stability data (ΔΔGua/kcal mol−1) provided where quantitative data is available
represents uncommon amino acid Gla. Coiled coil stability data (ΔΔGua/kcal mol−1) provided where quantitative data is available
| a ΔΔGu is the change in free energy of unfolding on addition of LaCl3. LaCl3 concentration for E2 and E3 peptides was 50 mM, and for Gla2Nx was 5 mM. A positive ΔΔGu indicates an increase in coiled coil stability. |
|---|
|
Further work by Hodges and co-workers showed that the stabilising effect of La3+ addition on their coiled coils was highly dependent on the position of the Gln to Glu substitutions.112 Three peptides were made, (E2(15,20), E2(13,22) and E2(20,22), Table 1), with varying distances between the two repulsive Glu e and g residues. Addition of LaCl3 had a greater stabilising effect on E2(15,20) coiled coil formation than for E2(13,22), reflecting the reduction in the magnitude of Glu–Glu repulsion as the distance between them increases. Whilst, of the three peptides, the Glu residues are physically closest in the E2(20,22) coiled coil, addition of LaCl3 is instead destabilising due to the presence of a d position leucine (Leu). In the two stranded coiled coil, this bulky Leu side chain protrudes into the interhelical interface between the two Glu residues of the two peptide chains, interfering with the repulsive interaction between the two Glu residues, thus reducing relief provided by La3+ binding.
Substitution of a third Glu in place of a Gln residue in the E2(15,20) peptide to give E(13,15,20) and E(15,20,22) (Table 1) resulted in enhanced coiled coil stability on addition of trivalent La3+ regardless of whether the third Glu was in position 13 or 22. The resulting charge neutrality provides additional stabilisation, even if direct lanthanide coordination to all three Glu residues is not possible.
Lanthanide binding can be further enhanced by replacement of Glu residues by the uncommon amino acid γ-carboxyglutamic acid (Gla).113 The Gla side chain contains two carboxylic acid functional groups (compared to the one carboxylate group of the Glu side chain). Under physiological pH, Gla therefore carries a charge of −2 and twice as many potential O-donor atoms. Free Gla had previously been shown to bind Tb3+via the side chain carboxylate groups in a Ln(III):Gla = 1:2 ratio.114
As might be expected, the increased magnitude of the negative charge as a result of Glu → Gla substitution results in greater destabilisation of the coiled coil structure. When the valine (Val) residue in position 23 was substituted for asparagine (Asn) in the Glu → Gla analogue of peptide E2(15,20) to give Gla2Nx (Table 1), the peptide adopted a random coil formation. Such is the affinity of La3+ for the Gla ligand, that LaCl3 addition was capable of recovering coiled coil formation to 100% and increasing coiled coil stability by up to 5.1 kcal mol−1.
Gla2Nx was also the first example of a de novo designed coiled coil peptide scaffold that was reported to bind lanthanides other than La3+; in spite of its smaller ionic radius, Yb3+ was shown to have a similar affinity for the Gla ligand as La3+. These peptides were shown to bind preferentially to lanthanides over other biologically relevant metals (Mg2+, Ca2+ and Zn2+). Binding preference for La3+ over Ca2+ is particularly notable given the similarity of lanthanide coordination chemistry with that of Ca2+ and is accounted for by the greater charge density of the trivalent lanthanide ion.
These early studies demonstrated that lanthanide binding sites could be engineered at the interface of complex helical peptides, and that lanthanide binding can even induce formation of higher order structures. Lanthanide binding can be achieved using readily available amino acids as ligands, with Gla being a particularly high affinity ligand. Importantly, these external binding sites show measurable selectivity for lanthanides over biologically relevant metals (Mg2+, Ca2+ and Zn2+).
Moving the lanthanide binding site into the hydrophobic core
In native metalloproteins the majority of metal binding sites are located buried within the hydrophobic core of the protein scaffold where the surrounding architecture can control metal coordination chemistry as well as solvent and substrate access. Given the importance of coordinated solvent in the chemistry of lanthanide complexes, it is extremely appealing to engineer such a site within the hydrophobic coiled coil core where the coordination chemistry of the lanthanide (including secondary coordination sphere effects) can be more easily controlled. The first report of lanthanide binding within the hydrophobic core of a coiled coil involved a Gla layer within a three stranded coiled coil (Pep3, see Table 2).115 Pep3 features a Gla layer (in position a of heptad 3) with a ‘hole’ generated by an Ala layer, directly above (position d of heptad 2) to better accommodate the bulk of the Gla side chain. Pep3 was found to bind a lanthanide ion (Eu3+, Ce3+ or Tb3+) whilst at the same time inducing helix formation. Pep4 and Pep5 were designed to self-assemble into a heterotrimeric coiled coil on addition of lanthanide ions.
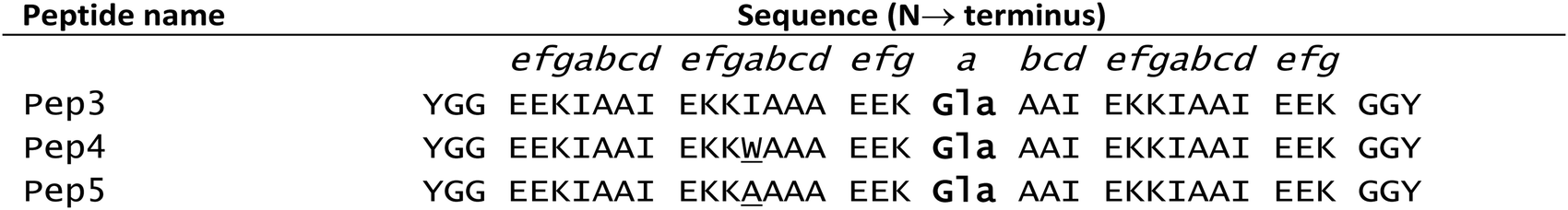
|
These peptides featured the Gla for lanthanide binding whilst Pep4 also contains a tryptophan (Trp) as a sensitiser for lanthanide luminescence in position a of heptad 2 (i.e. in the layer above the Gla site), and Pep5 contained an Ala as the complementary ‘hole’ to accommodate the bulk of the Trp. Addition of either Eu3+, Tb3+ or Ce3+ induced formation of a trimeric coiled coil Ln(Pep4)(Pep5)2 and was accompanied by Trp sensitised Ln luminescence.
This work demonstrated for the first time that lanthanide binding sites could be engineered within the hydrophobic core of a coiled coil. However, the focus was predominantly on the effect of lanthanide ions on peptide folding and further exploration of the lanthanide coordination chemistry or their photophysical properties was not reported.
Lanthanide binding sites using common amino acids
The use of commonly occurring amino acids holds great appeal as the use of non-natural, or less common amino acids can be expensive and tends to restrict their use to synthetically accessible peptide sequences. Restricting use to the 20 common amino acids, it was possible for the first time to generate a lanthanide binding site within the hydrophobic core of a coiled coil using aspartic acid (Asp) and Asn.116Peptide design was based on a repeating heptad of Ac-G(IAAIEQK)xG-NH2 which favours three-stranded coiled coil formation. The lanthanide binding site was rationally designed based around the coordination of Asp and Asn side chains. Asp was preferred to Glu because initial molecular dynamics modelling of the binding site suggested appropriate Ln–O bond lengths for the Asp side chain. Coordination of a Ln3+ ion to the Asp side chains in the core of a trimeric coiled coil will therefore lead to charge neutrality. The a site Asp (heptad 3) would also provide up to six oxygen donor atoms at roughly the desired distance for Ln–O bond formation. The Asn residue was located directly above the Asp layer (the d position of the second heptad) to provide an additional layer of oxygen donor atoms, thereby providing the Ln3+ with up to nine oxygen donor atoms. Finally, a Trp residue, a known sensitiser for some Ln3+ ions, was introduced adjacent to the designed binding site in an f position (MB1, see Table 3). Modelling of the design predicted binding of Ln3+ ions via three Asp side chains and three carbonyl oxygens of the Asn residues as well as the presence of a directly bound water molecule (Fig. 3). As was previously observed for lanthanide coiled coils, in the absence of any coordinating metal, the negatively charged coordinating side chains repel each other and destabilise the coiled coil structure. However, titration of Ln3+ ions into a solution of MB1 peptide at pH 7.0 induced folding reaching maximum folding at one equivalent of Ln3+ per peptide trimer, consistent with the design. Similar behaviour was observed from titrating a range of different lanthanide ions, (Ce3+, Nd3+, Eu3+, Dy3+, Er3+ and Yb3+) including the first report of a Gd3+ coiled coil complex.
 where available. Metal-binding residues are highlighted in bold and the sensitising Trp or Tyr residues are underlined. Peptides form trimeric coiled coils. For peptides MB1-1(2X), X = alanine (A), isoleucine (I), or phenylalanine (F)
where available. Metal-binding residues are highlighted in bold and the sensitising Trp or Tyr residues are underlined. Peptides form trimeric coiled coils. For peptides MB1-1(2X), X = alanine (A), isoleucine (I), or phenylalanine (F)


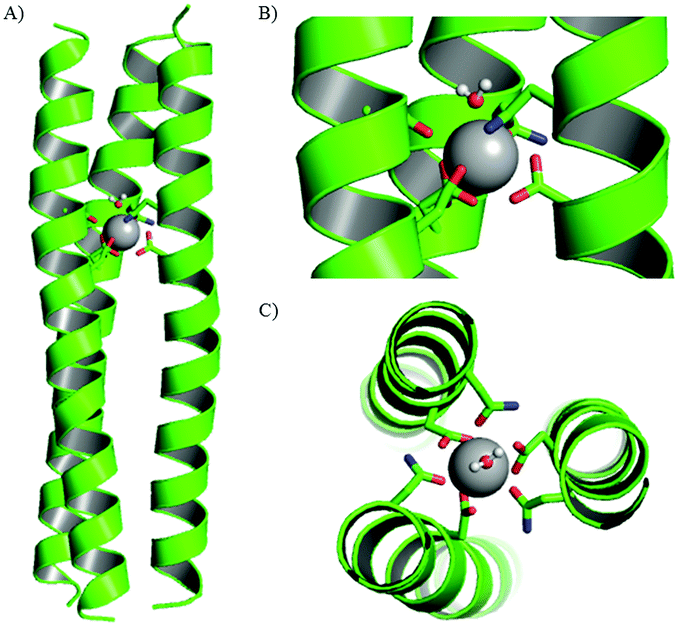 | ||
| Fig. 3 (A) Structure of Gd(MB1)3 after 10.0 ns of molecular dynamics simulations and close up, (B) side-on and (C) top-down views of the Gd3+ coordination site. Shown are the main chain atoms represented as helical ribbons (green) and the Asn and Asp side chains in stick form (oxygen in red and nitrogen in blue), a water molecule in ball-and-stick form and the Gd3+ ion as a sphere in grey. Reproduced from ref. 116 (https://pubs.acs.org/doi/10.1021/ja408741h) with permission from the American Chemical Society. Permission for further use of this figure must be sought from the American Chemical Society. | ||
As with previously reported lanthanide-binding coiled coils, the binding site was specific for Ln3+ ions over Ca2+ ions. This is most likely a result of the complementary charge within a trimeric coiled coil. However, contrary to the model's prediction, investigation into the peptide-bound Tb3+ hydration state showed no water molecules were directly coordinated. Surprisingly, despite the lack of inner sphere water, MRI properties of Gd(MB1)3 were superior to those of Gd(DOTA) which is in clinical use as an MRI contrast agent under the trade name Dotarem. Gd(MB1)3 showed >three-fold enhanced transverse relaxivity, r2, at 7 T in comparison to Gd(DOTA).
The superior MRI relaxivities of this gadolinium coiled coil, achieved despite the lack of inner sphere water, which is normally considered a prerequisite for gadolinium MRI contrast agents, were the motivation to explore more fully the opportunities afforded by this new class of ligands for the lanthanide ions, and gadolinium in particular.
The following sections detail our efforts to tune the chemistry of lanthanide ions coordinated to coiled coil ligands, through modifications to both primary and secondary coordination sphere ligands. Furthermore, the size of these coiled coil ligands provides an opportunity for the inclusion of multiple distinct metal binding sites within a single design.
Linear translation of a lanthanide binding site along a coiled coil
MRI relaxivity, a measure of contrast agent efficiency, tends to correlate with the number of gadolinium-bound water molecules, as increasing this should improve efficiency of transfer of magnetisation to the bulk solvent. Whilst experimental work showed no water was directly bound to the MB1-bound Tb3+, the molecular dynamics modelling indicated that this was not necessarily due to lack of physical space at the metal binding site, but may rather reflect the fact that the site is located centrally, buried within the coiled coil hydrophobic core. Thus, binding sites located closer to the peptide termini may be more amenable to water access.To investigate this, the binding site, including the adjacent Trp sensitiser, was systematically translated along the coiled coil (Fig. 4 and Table 3).117 The Asn3Asp3 metal binding site in MB1 is situated between the second and third heptad. However, there are three other sites in which this binding site could reside, one a heptad above and two in lower heptads. These peptides were denoted MB1-x where x = 1–4 and refers to the heptad containing the Asn residue, with the Asp residue in the a site of the following heptad. The original MB1 peptide is therefore renamed MB1-2.
 | ||
| Fig. 4 Relationship between linear location of Asn3Asp3 Ln3+ binding site and coordination chemistry. Shown from left to right are cartoons of (A) MB1-1, (B) MB1-2, (C) MB1-3 and (D) MB1-4. Shown are the main chain atoms represented as helical ribbons (green), the Asn and Asp side chains in stick form (oxygen in red and nitrogen in blue) and the Gd3+ ion as a sphere (pink). Reproduced from ref. 117 with permission from the Royal Society of Chemistry. | ||
Core heptads have been reported to be up to three times more stabilising with respect to coiled coil formation than terminal heptads.118 Consistent with this observation, peptides with lanthanide binding sites located in core heptads were considerably less folded than peptides containing these in terminal heptads. Of the two peptides containing terminal binding sites, MB1-1 at the N-terminus and MB1-4 at the C-terminus, MB1-1 was both better folded and more stable. This is because the coiled coil is not symmetrical and the MB1-1 and MB1-4 sites are not identical. The binding site in MB1-1 is closer to the extremity of the coiled coil, whereas it is more buried in MB1-4. A similar trend was seen in the folding of the Gd-peptides, Gd(MB1-1)3 > Gd(MB1-4)3 > Gd(MB1-2)3 ∼ Gd(MB1-3)3.
Regardless of binding site location, the emission profile of bound Eu3+ in Eu(MB1-X)3 indicated a symmetric Eu3+ site, consistent with a single Eu3+ bound within a symmetric coiled coil. In contrast, the hydration state of bound Tb3+ was found to be highly dependent on the binding site location. As in the previous study, there was no evidence of inner sphere water when Tb3+ is bound centrally along the coiled coil (MB1-2 and MB1-3). However, water was able to directly coordinate to Tb3+ when bound in terminal binding sites, with evidence of three and two inner sphere waters for MB1-1 and MB1-4, respectively. Not surprisingly, the increase in inner sphere water correlates with an increase in MRI relaxivity for the Gd3+ analogues.
Gd(MB1-2)3 and Gd(MB1-3)3, with no evidence of inner sphere water, display comparable transverse and longitudinal relaxivity. Increasing the hydration of the bound metal to two (Gd(MB1-4)3) and three (Gd(MB1-1)3) water molecules increases the relaxivity accordingly. Consequently the 1 nm linear translation of an otherwise identical ligand presenting binding site along the coiled coil has altered the water access and lanthanide coordination chemistry to such an extent that we observe a four-fold increase in MRI transverse relaxivity of the Gd3+ complex.
The lack of inner sphere water for lanthanides coordinated to the centrally located binding sites is consistent with a coordinatively saturated environment where the peptide scaffold provides all of the coordinating donor atoms. However, the lanthanide hydration states at the terminal binding sites suggest that not all of the Asp/Asn residues are fully engaged in metal binding. In the case of MB1-1, it was proposed that the Asn layer does not coordinate to the bound lanthanide. To test this hypothesis, the analogous peptide which lacked the Asn layer, CS1-1, (Table 3), was prepared. This design was still able to bind lanthanide ions, and had a similar experimentally observed hydration state to MB1-1, suggesting the Asn residue was not essential for metal binding at the C-terminus.
CS1-1 was found to display optimal Tb3+ binding between pH 6–7. At low pH, it was speculated that protonation of Asp side chains precludes lanthanide binding, whilst at high pH, competing formation of lanthanide hydroxide species causes dissociation of Tb3+. Similar observations were made for the MB1-2 peptide, although the pH range through which Tb3+ was bound was considerably larger (pH 4–7).119
Secondary coordination sphere effects
Given that natural metalloproteins very effectively control coordination chemistry with secondary coordination sphere effects, this presents a strategy that can be adopted for tuning the coordination chemistry achieved with coiled coil ligands. Changes to the identities of non-coordinating residues within a coiled coil has previously been shown to influence various aspects of metal coordination chemistry. For example, changing the steric bulk of residues situated in an adjacent layer to the metal binding site, can influence the number of exogenous ligands around the metal.95,120–124 Using these principles, steric changes to the secondary coordination sphere have also been used to tune the hydration state of bound lanthanide ions.125The starting point was the binding site within MB1-1, located towards the N-terminus of the coiled coil, which generates a highly hydrated lanthanide site, Tb(OH2)3(MB1-1)3. MB1-1, contains a non-coordinating terminal isoleucine (Ile) layer directly above the Asn/Asp binding site residues (in the a position of the first heptad) and it was the identity of this residue that was systematically altered to Ala, phenylalanine (Phe), tyrosine (Tyr) and Trp (MB1-1(2X), Fig. 5 and Table 3).
 | ||
| Fig. 5 Pymol models of the MB1-1(2X) peptide illustrating mutation of the residue in position two (2X) through a space filling representation and their influence on lanthanide hydration state. The main chain atoms are represented as helical ribbons (green), Asn and Asp coordinating residues along with the modified residue in position two (2X) are shown in stick form (nitrogen in blue and oxygen in red) and the Ln3+ ion as a grey sphere. Top down view of the proposed coodinatively saturated lanthanide binding site. Reproduced from ref. 125 with permission from the Royal Society of Chemistry. | ||
Ala is sterically less bulky than Ile, and so might have been expected to increase the hydration state by allowing greater water access. However, luminescence lifetime decay experiments featuring Tb3+ as the bound lanthanide showed that the hydration state remained three, indicating that for the MB1-1 Asn/Asp binding site, this is the maximum hydration state accessible. In contrast, Phe which is bulkier than Ile leads to a less hydrated site with two water molecules bound to the Tb3+. As the steric bulk of the terminal second sphere residue increased further, so did the reduction in hydration state. Introduction of Tyr resulted in Tb3+ bound to the Asn/Asp site with one exogenous water molecule, and, with the bulkier Trp, water binding was prevented completely. Introduction of this second coordination sphere Trp therefore converts the terminal binding site into one more closely resembling a buried binding site located centrally within the coiled coil (MB1-2 or MB1-3), and this is mirrored in the chemistry of the sites, including near identical MRI relaxivity data for the Gd3+ complexes of MB1-1(2W) and MB1-2. Altering the identity of a single, second coordination sphere terminal residue in MB1-1, from Ile to the bulkier Trp, results in a four-fold reduction in transverse relaxivity for the Gd3+ complex.
Heterometallic coiled coils containing d- and f-block metals
Engineering multinuclear systems containing distinct mononuclear sites has great appeal. Within such systems, the distinct properties of the different metals, or the same metal but with different coordination chemistry, may be harnessed either independently from each other or in a complementary fashion. Indeed, nature makes use of such systems in heteronuclear metalloenzymes such as heme-copper oxidase and cytochrome c oxidase.126–128 As such, the rational design of multinuclear coiled coils with distinct, controlled coordination environments presents opportunities for the synthesis of multifunctional and potentially responsive systems. For example, second metal binding sites may act to enhance the properties of mononuclear systems by providing additional stability or via preorganisation of active sites. Pecoraro and co-workers have designed a number of heterometallic coiled coils,93,95,129 including those featuring both structural and catalytic metal sites.87,94 Another example of a hetero-bimetallic coiled coil incorporated a Ru(bipyridyl)3 site at one end which preorganised the scaffold, enabling copper binding to a His3 site at the opposite end.130Only one example of a lanthanide-containing hetero-bimetallic coiled coil has thus far been reported.119 The coiled coil features a lanthanide binding Asp3 site as well as a thiolate Cys3 site, suitable for binding softer metal ions including mercury.90,131 The soft thiol ligands contrast with the hard oxygen donors associated with lanthanide-binding, and provide a mechanism by which selectivity can be readily achieved based on hard-soft acid base theory. The presence of polar metal binding residues in the hydrophobic core of a coiled coil is destabilising and so both binding sites were located at opposite termini so as to minimise destabilisation.117 The Cys3 mercury binding site was located in the a position of the fifth, a terminal, heptad; a site that has been previously reported and well-studied.90,131 The least disruptive of the lanthanide binding sites studied to date, CS1-1, was located at the N-terminus and contains a single Asp layer in the a position of the second heptad. Thus the designed hetero-bimetallic coiled coil contained an Asp3 layer for lanthanide binding towards the N-terminus and a Cys3 site for mercury binding ∼4 nm away towards the C-terminus, to generate CS2-1,4 (Fig. 6).
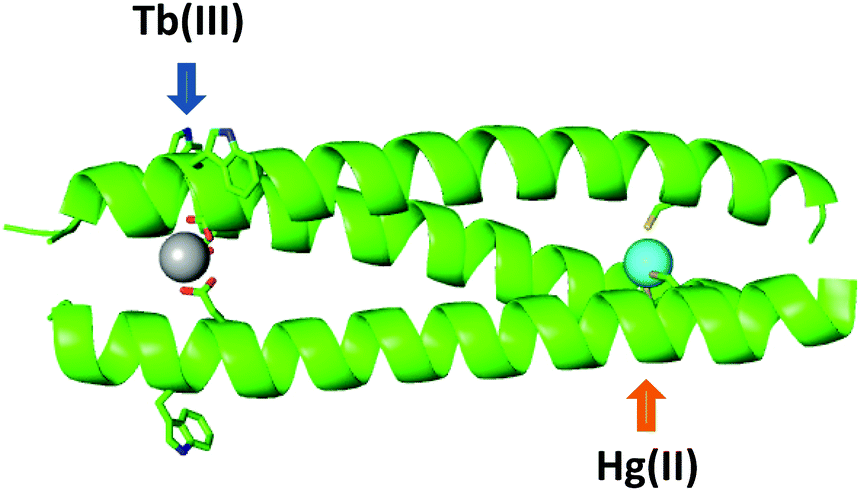 | ||
| Fig. 6 Cartoon representation showing designed hetero bimetallic coiled coil, CS2-1,4, featuring LnAsp3 and HgCys3 sites towards the N- and C-termini, respectively. Shown are the main chain atoms represented as helical ribbons (green), the Asp, Trp and Cys side chains in stick form (oxygen in red, nitrogen in blue and sulfur in orange), and the metal ions as spheres (lanthanide in grey and mercury in cyan). Reproduced from ref. 119 with permission from the Royal Society of Chemistry. | ||
Mercury binding to the Cys3 site is known to be pH dependent, with binding as HgCys2 at neutral pH changing to HgCys3 above pH 7.6.132 The same behaviour was seen in the CS2-1,4 peptide, demonstrating that Hg2+ binding was selective for the soft Cys3 site and was insensitive to the presence of the hard Asp3 binding site. Luminescence experiments also showed that, at neutral pH, Tb3+ binding to CS2-1,4 was similar to Tb3+ binding to peptides lacking the Cys3 mercury binding site. Again, binding is selective for the intended, in this case hard Ln3+, metal binding site with no interference due to the presence of a second Cys3 metal binding site.
In this hetero-bimetallic system both d- and f-block metal binding sites were successfully introduced within a single coiled coil, demonstrating that it should be possible to exploit their complementary attractive properties. These could include the photophysical and magnetic properties of lanthanide sites, coupled with the breadth of electronic, catalytic or spectroscopic properties afforded by the d-block metals. Secondly the d- and f-block metal binding sites operated independently from each other, showing essentially the same binding behaviour as in the corresponding mononuclear peptides. As such, future design can select from an extensive toolbox of designed monometallic sites, in a “plug-and-play” type approach to achieve functional complex designs.
Outlook and conclusions
Far from their early description of ‘boring’,1 lanthanides have become key to many technologies. Components in microchips, catalytic converters, magnets and lasers, lanthanides are ubiquitous in modern life.133 The magnetic and optical properties of the lanthanides have also made them highly useful as imaging probes,2 including as MRI contrast agents5 and luminescent sensors. Lanthanides have also proven to be useful tools in probing the structure and function of proteins.4,67 The recent discovery of native biochemical roles associated with lanthanides6,7 further underscores the importance of understanding lanthanide–protein assemblies and presents further opportunities to develop these for new technologies, for example as a means to detect and extract lanthanides from their naturally occurring ores.70As with other metals, the coordination environment has been shown to play an essential role in the activity and behaviour of lanthanide complexes.102–104 Thus, if the true potential of lanthanide–protein assemblies is to be realised, the ability to rationally design lanthanide binding sites with predictable chemistry is essential. Coiled coils are a class of ligands which benefit from many of the advantages afforded by protein ligands without much of the complexity. Rational design principles can be used to tune the lanthanide coordination chemistry.
One challenge that remains is to improve the binding affinities of lanthanides to the coiled coil scaffolds. Table 4 provides a summary of lanthanide-binding coiled coils discussed in this review, with their principle metal-binding residues and logK values (where data was available). For comparison, Gd-DOTA, a small molecule MRI contrast agent in clinical use, has a logK of ∼26, whereas lanthanide binding tags have logK values of ∼8–9, and the lanthanide coiled coils typically have logK values of ∼5. To use coiled coil-based systems for in vivo applications, significant improvements to binding affinity would be necessary. However, for in vitro, assay-based diagnostics these binding affinities may be adequate.
K) where available
| Peptide | Principle metal binding residues | Lanthanides investigated | Binding affinity (logK) |
|---|---|---|---|
|
a logK values calculated from approximate dissociation constants (Kd) given in original work.112
b logK values and errors calculated from dissociation constants (Kd) and errors given in original work.113
c Quantitative data on lanthanide binding not available.
d logK values quoted directly from the original work.117
|
|||
| Ln(E2(15,20))2 | Glu | La3+ | ∼1.9 (La3+)a |
| Ln(E3(13,15,20))2 | Glu | La3+ | ∼2.5 (La3+)a |
| Ln(E3(15,20,22))2 | Glu | La3+ | N/A |
| Ln(Gla2Nx)2 | Gla | La3+, Yb3+ | 6.2 ± 0.2 (La3+), 6.4 ± 0.2 (Yb3+)b |
| Ln(Pep3)3 | Gla | Ce3+, Nd3+, Eu3+, Dy3+, Er3+ and Yb3+ | N/Ac |
| Ln(Pep4)(Pep5)2 | Gla | Ce3+, Eu3+, Tb3+ | N/Ac |
| Ln(MB1-1)3 | Asp, Asn | Eu3+, Gd3+, Tb3+ | 5.30 ± 0.15 (Tb3+)d |
| Ln(MB1-2)3 | Asp, Asn | Ce3+, Nd3+, Eu3+, Gd3+, Tb3+, Dy3+, Er3+ and Yb3+ | 5.48 ± 0.20 (Tb3+)d |
| Ln(MB1-3)3 | Asp, Asn | Eu3+, Gd3+, Tb3+ | 5.16 ± 0.26 (Tb3+)d |
| Ln(MB1-4)3 | Asp, Asn | Eu3+, Gd3+, Tb3+ | 5.26 ± 0.36 (Tb3+)d |
| Ln(CS1-1)3 | Asp | Tb3+ | 4.57 ± 0.07 (Tb3+)d |
The potential of the coiled coil scaffold lies in the demonstrated ability to control coordination chemistry by making simple, rational changes to peptide sequence. For example, the lanthanide coordination environments for MRI contrast agents versus luminescent probes have different requirements. To optimise lanthanide luminescence, a sensitising ‘antenna’ (usually an aromatic organic group) must be incorporated, and water must be excluded from the coordination sphere to prevent quenching. Conversely, for MRI contrast agents, increasing the number of coordinating water molecules is desirable as this correlates with increased relaxivity. The ability to fine tune the lanthanide coordination environment to predictably either bind or exclude water means that the coiled coil is a versatile scaffold potentially capable of sensitising lanthanide luminescence and also acting as an MRI contrast agent, depending on the design of the lanthanide binding site. As such, this new class of lanthanide complexes provide valuable insights into the relationship between coordination environment and lanthanide complex behaviour and are therefore an important addition to the toolbox for scientists seeking to design new functional lanthanide-protein assemblies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the School of Chemistry and the University of Birmingham for support of this work.References
- G. C. Pimentel and R. D. Spratley, Understanding Chemistry, Holden-Day Inc., San Fransisco, 1971, p. 862 Search PubMed.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 Search PubMed.
- B. C. Mayo, Chem. Soc. Rev., 1973, 2, 49–74 Search PubMed.
- K. N. Allen and B. Imperiali, Curr. Opin. Chem. Biol., 2010, 14, 247–254 Search PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 Search PubMed.
- L. J. Daumann, Angew. Chem., Int. Ed., 2019, 58, 12795–12802 Search PubMed.
- J. A. Cotruvo, ACS Cent. Sci., 2019, 5, 1496–1506 Search PubMed.
- V. Putignano, A. Rosato, L. Banci and C. Andreini, Nucleic Acids Res., 2018, 46, D459–D464 Search PubMed.
- R. L. Shook and A. S. Borovik, Inorg. Chem., 2010, 49, 3646–3660 Search PubMed.
- S. A. Cook, E. A. Hill and A. S. Borovik, Biochemistry, 2015, 54, 4167–4180 Search PubMed.
- S. A. Cook and A. S. Borovik, Acc. Chem. Res., 2015, 48, 2407–2414 Search PubMed.
- R. L. Shook and A. S. Borovik, Chem. Commun., 2008, 6095–6107 Search PubMed.
- S. I. Mann, T. Heinisch, T. R. Ward and A. S. Borovik, Chem. Commun., 2018, 54, 4413–4416 Search PubMed.
- L. R. Marshall, O. Zozulia, Z. Lengyel-Zhand and I. V. Korendovych, ACS Catal., 2019, 9, 9265–9275 Search PubMed.
- M. Winkler, M. Geier, S. P. Hanlon, B. Nidetzky and A. Glieder, Angew. Chem., Int. Ed., 2018, 57, 13406–13423 Search PubMed.
- K. M. Koeller and C.-H. Wong, Nature, 2001, 409, 232–240 Search PubMed.
- H.-P. Meyer, E. Eichhorn, S. Hanlon, S. Lütz, M. Schürmann, R. Wohlgemuth and R. Coppolecchia, Catal. Sci. Technol., 2013, 3, 29–40 Search PubMed.
- G. A. Strohmeier, H. Pichler, O. May, M. Gruber-Khadjawi, G. A. Strohmeier, H. Pichler, O. May and M. Gruber-Khadjawi, Chem. Rev., 2011, 111, 4141–4164 Search PubMed.
- G. Hughes and J. C. Lewis, Chem. Rev., 2018, 118, 1–3 Search PubMed.
- G. Platt, C.-W. Chung and M. S. Searle, Chem. Commun., 2001, 1162–1163 Search PubMed.
- R. P. Bonomo, L. Casella, L. De Gioia, H. Molinari, G. Impellizzeri, T. Jordan, G. Pappalardo, R. Purrello and E. Rizzarelli, J. Chem. Soc., Dalton Trans., 1997, 2387–2390 Search PubMed.
- J.-C. G. Bünzli, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, Amsterdam, 2016, vol. 50, ch. 287, pp. 141–176 Search PubMed.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 Search PubMed.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 Search PubMed.
- L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 Search PubMed.
- A. K. R. Junker, L. R. Hill, A. L. Thompson, S. Faulkner and T. J. Sørensen, Dalton Trans., 2018, 47, 4794–4803 Search PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. Gareth Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 Search PubMed.
- F. Martelli, S. Abadie, J.-P. Simonin, R. Vuilleumier and R. Spezia, Pure Appl. Chem., 2012, 85, 237–246 Search PubMed.
- S. A. Cotton, C. R. Chim., 2005, 8, 129–145 Search PubMed.
- E. N. Rizkalla and G. R. Choppin, in Handbook on the Physics and Chemistry of the Rare Earths, ed. K. A. Gschneidner Jr., L. Eyring, G. R. Chopping and G. H. Lander, Elsevier, Amsterdam, 1994, vol. 18, ch. 127, pp. 529–558 Search PubMed.
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1–271 Search PubMed.
- A. F. Cockerill, G. L. O. Davies, R. C. Harden and D. M. Rackham, Chem. Rev., 2002, 73, 553–588 Search PubMed.
- T. Saio and F. Inagaki, in Advanced Methods in Structural Biology, ed. T. Senda and K. Maenaka, Springer, Tokyo, 2016, ch. 17, pp. 315–340 Search PubMed.
- I. Bertini, P. Kursula, C. Luchinat, G. Parigi, J. Vahokoski, M. Wilmanns and J. Yuan, J. Am. Chem. Soc., 2009, 131, 5134–5144 Search PubMed.
- I. Bertini, Y. K. Gupta, C. Luchinat, G. Parigi, M. Peana, L. Sgheri and J. Yuan, J. Am. Chem. Soc., 2007, 129, 12786–12794 Search PubMed.
- I. Bertini, C. Luchinat, G. Parigi and R. Pierattelli, Dalt. Trans, 2008, 3782–3790 Search PubMed.
- J. Lux and A. D. Sherry, Curr. Opin. Biol. Chem., 2018, 45, 121–130 Search PubMed.
- J. Garcia, S. Z. Liu and A. Y. Louie, Philos. Trans. R. Soc., A, 2017, 375, 20170180 Search PubMed.
- S. Hirano and K. T. Suzuki, Environ. Health Perspect., 1996, 104, 85–95 Search PubMed.
- J. C. Bousquet, S. Saini, D. D. Stark, P. F. Hahn, M. Nigam, J. Wittenberg and J. T. Ferrucci, Radiology, 1988, 166, 693–698 Search PubMed.
- P. J. Klemm, W. C. Floyd III, D. E. Smiles, J. M. J. Fréchet and K. N. Raymond, Contrast Media Mol. Imaging, 2012, 7, 95–99 Search PubMed.
- A. Koudrina and M. C. DeRosa, ACS Omega, 2020, 5, 22691–22701 Search PubMed.
- S. Lim and S. J. Franklin, Cell. Mol. Life Sci., 2004, 61, 2184–2188 Search PubMed.
- Y. Hibi, K. Asai, H. Arafuka, M. Hamajima, T. Iwama and K. Kawai, J. Biosci. Bioeng., 2011, 111, 547–549 Search PubMed.
- A. Pol, T. R. M. Barends, A. Dietl, A. F. Khadem, J. Eygensteyn, M. S. M. Jetten and H. J. M. Op den Camp, Environ. Microbiol., 2014, 16, 255–264 Search PubMed.
- M. Taubert, C. Grob, A. M. Howat, O. J. Burns, J. L. Dixon, Y. Chen and J. C. Murrell, Environ. Microbiol., 2015, 17, 3937–3948 Search PubMed.
- M. C. Macey, J. Pratscher, A. T. Crombie and J. C. Murrell, Microbiome, 2020, 8, 31 Search PubMed.
- A. M. Ochsner, L. Hemmerle, T. Vonderach, R. Nüssli, M. Bortfeld-Miller, B. Hattendorf and J. A. Vorholt, Mol. Microbiol., 2019, 111, 1152–1166 Search PubMed.
- L. Chistoserdova, World J. Microbiol. Biotechnol., 2016, 32, 138 Search PubMed.
- N. Picone and H. J. M. Op den Camp, Curr. Opin. Chem. Biol., 2019, 49, 39–44 Search PubMed.
- J. L. Gifford, M. P. Walsh and H. J. Vogel, Biochem. J., 2007, 405, 199–221 Search PubMed.
- C.-L. A. Wang, R. R. Aquaron, P. C. Leavis and J. Gergely, Eur. J. Biochem., 1982, 124, 7–12 Search PubMed.
- J. Hu, X. Jia, Q. Li, X. Yang and K. Wang, Biochemistry, 2004, 43, 2688–2698 Search PubMed.
- H. G. Brittain, F. S. Richardson, R. B. Martin, H. G. Brittain, F. S. Richardson and R. Bruce Martin, J. Am. Chem. Soc., 1976, 98, 8255–8260 Search PubMed.
- C. H. Evans, Trends Biochem. Sci., 1983, 8, 445–449 Search PubMed.
- J. P. MacManus, C. W. Hogue, B. J. Marsden, M. Sikorska and A. G. Szabo, J. Biol. Chem., 1990, 265, 10358–10366 Search PubMed.
- K. J. Franz, M. Nitz and B. Imperiali, ChemBioChem, 2003, 4, 265–271 Search PubMed.
- M. Nitz, K. J. Franz, R. L. Maglathlin and B. Imperiali, ChemBioChem, 2003, 4, 272–276 Search PubMed.
- L. J. Martin, B. R. Sculimbrene, M. Nitz and B. Imperiali, QSAR Comb. Sci., 2005, 24, 1149–1157 Search PubMed.
- M. Nitz, M. Sherawat, K. J. Franz, E. Peisach, K. N. Allen and B. Imperiali, Angew. Chem., Int. Ed., 2004, 43, 3682–3685 Search PubMed.
- A. W. Barb and G. P. Subedi, J. Biomol. NMR, 2016, 64, 75–85 Search PubMed.
- K. Barthelmes, A. M. Reynolds, E. Peisach, H. R. A. Jonker, N. J. DeNunzio, K. N. Allen, B. Imperiali and H. Schwalbe, J. Am. Chem. Soc., 2010, 133, 808–819 Search PubMed.
- N. R. Silvaggi, L. J. Martin, H. Schwalbe, B. Imperiali and K. N. Allen, J. Am. Chem. Soc., 2007, 129, 7114–7120 Search PubMed.
- T. Zhuang, H.-S. Lee, B. Imperiali and J. H. Prestegard, Protein Sci., 2008, 17, 1220–1231 Search PubMed.
- L. J. Martin, M. J. Hähnke, M. Nitz, J. Wöhnert, N. R. Silvaggi, K. N. Allen, H. Schwalbe and B. Imperiali, J. Am. Chem. Soc., 2007, 129, 7106–7113 Search PubMed.
- B. R. Sculimbrene and B. Imperiali, J. Am. Chem. Soc., 2006, 128, 7346–7352 Search PubMed.
- T. W. Victor, K. H. O’Toole, L. M. Easthon, M. Ge, R. J. Smith, X. Huang, H. Yan, Y. S. Chu, S. Chen, D. Gursoy, M. Ralle, B. Imperiali, K. N. Allen and L. M. Miller, J. Am. Chem. Soc., 2020, 142, 2145–2149 Search PubMed.
- K. D. Daughtry, L. J. Martin, A. Sarraju, B. Imperiali and K. N. Allen, ChemBioChem, 2012, 13, 2567–2574 Search PubMed.
- J. A. Cotruvo, E. R. Featherston, J. A. Mattocks, J. V. Ho, T. N. Laremore, J. A. Cotruvo, E. R. Featherston, J. A. Mattocks, J. V. Ho and T. N. Laremore, J. Am. Chem. Soc., 2018, 140, 15056–15061 Search PubMed.
- J. A. Mattocks, J. V. Ho and J. A. Cotruvo, J. Am. Chem. Soc., 2019, 141, 2857–2861 Search PubMed.
- S. J. Caldwell, I. C. Haydon, N. Piperidou, P.-S. Huang, M. J. Bick, H. S. Sjöström, D. Hilvert, D. Baker and C. Zeymer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30362 LP Search PubMed.
- C. S. Bonnet, P. H. Fries, S. Crouzy, O. Sénèque, F. Cisnetti, D. Boturyn, P. Dumy and P. Delangle, Chem. – Eur. J., 2009, 15, 7083–7093 Search PubMed.
- C. S. Bonnet, P. H. Fries, S. Crouzy and P. Delangle, J. Phys. Chem. B, 2010, 114, 8770–8781 Search PubMed.
- M. Isaac, A. Pallier, F. Szeremeta, P.-A. Bayle, L. Barantin, C. S. Bonnet and O. Sénèque, Chem. Commun., 2018, 54, 7350–7353 Search PubMed.
- M. Isaac, L. Raibaut, C. Cepeda, A. Roux, D. Boturyn, S. V. Eliseeva, S. Petoud and O. Sénèque, Chem. – Eur. J., 2017, 23, 10992–10996 Search PubMed.
- C. W. am Ende, H. Y. Meng, M. Ye, A. K. Pandey and N. J. Zondlo, ChemBioChem, 2010, 11, 1738–1747 Search PubMed.
- L. L. Liu and K. J. Franz, J. Am. Chem. Soc., 2005, 127, 9662–9663 Search PubMed.
- L. L. Liu and K. J. Franz, JBIC, J. Biol. Inorg. Chem., 2007, 12, 234–247 Search PubMed.
- L. Ancel, A. Niedźwiecka, C. Lebrun, C. Gateau and P. Delangle, C. R. Chim., 2013, 16, 515–523 Search PubMed.
- A. J. Gamble and A. F. A. Peacock, in Methods in Molecular Biology, 2nd edn, ed. V. Köhler, Humana Press, New York, 2014, vol. 1216, ch. 11, pp. 211–231 Search PubMed.
- A. J. Doerr and G. L. McLendon, Inorg. Chem., 2004, 43, 7916–7925 Search PubMed.
- J. S. Plegaria and V. L. Pecoraro, Isr. J. Chem., 2015, 55, 85–95 Search PubMed.
- C. S. Mocny and V. L. Pecoraro, Acc. Chem. Res., 2015, 48, 2388–2396 Search PubMed.
- A. Lombardi, F. Pirro, O. Maglio, M. Chino and W. F. DeGrado, Acc. Chem. Res., 2019, 52, 1148–1159 Search PubMed.
- M. Y. Ogawa, J. Fan, A. Fedorova, J. Hong, O. A. Kharenko, A. Y. Kornilova, R. C. Lasey and F. Xie, J. Braz. Chem. Soc., 2006, 17, 1516–1521 Search PubMed.
- D. Ghosh and V. L. Pecoraro, Inorg. Chem., 2004, 43, 7902–7915 Search PubMed.
- M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Nat. Chem., 2012, 4, 118–123 Search PubMed.
- J. Grzyb, F. Xu, L. Weiner, E. J. Reijerse, W. Lubitz, V. Nanda and D. Noy, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 406–413 Search PubMed.
- K. Suzuki, H. Hiroaki, D. Kohda, H. Nakamura and T. Tanaka, J. Am. Chem. Soc., 1998, 120, 13008–13015 Search PubMed.
- G. R. Dieckmann, D. K. McRorie, D. L. Tierney, L. M. Utschig, C. P. Singer, T. V. O’Halloran, J. E. Penner-Hahn, W. F. DeGrado and V. L. Pecoraro, J. Am. Chem. Soc., 1997, 119, 6195–6196 Search PubMed.
- X. Li, K. Suzuki, A. Kashiwada, H. Hiroaki, D. Kohda and T. Tanaka, Protein Sci., 2000, 9, 1327–1333 Search PubMed.
- T. Tanaka, T. Mizuno, S. Fukui, H. Hiroaki, J. Oku, K. Kanaori, K. Tajima and M. Shirakawa, J. Am. Chem. Soc., 2004, 126, 14023–14028 Search PubMed.
- A. E. Tolbert, C. S. Ervin, L. Ruckthong, T. J. Paul, V. M. Jayasinghe-Arachchige, K. P. Neupane, J. A. Stuckey, R. Prabhakar and V. L. Pecoraro, Nat. Chem., 2020, 12, 405–411 Search PubMed.
- M. L. Zastrow and V. L. Pecoraro, J. Am. Chem. Soc., 2013, 135, 5895–5903 Search PubMed.
- A. F. A. Peacock, L. Hemmingsen and V. L. Pecoraro, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16566–16571 Search PubMed.
- E. Haviv, D. Azaiza-Dabbah, R. Carmieli, L. Avram, J. M. L. Martin and R. Neumann, J. Am. Chem. Soc., 2018, 140, 12451–12456 Search PubMed.
- P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2019, 58, 4504–4509 Search PubMed.
- N. M. Marshall, D. K. Garner, T. D. Wilson, Y.-G. Gao, H. Robinson, M. J. Nilges and Y. Lu, Nature, 2009, 462, 113–116 Search PubMed.
- A. Lombardi, C. M. Summa, S. Geremia, L. Randaccio, V. Pavone and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6298–6305 Search PubMed.
- K. J. Koebke, F. Yu, E. Salerno, C. Van Stappen, A. G. Tebo, J. E. Penner-Hahn and V. L. Pecoraro, Angew. Chem., Int. Ed., 2018, 57, 3954–3957 Search PubMed.
- B. Therrien, Front. Chem., 2018, 6, 602 Search PubMed.
- N. E. Borisova, T. B. Sumyanova, A. V. Kharcheva, P. I. Matveev, A. V. Ivanov, E. A. Razumova and S. V. Patsaeva, Dalton Trans., 2018, 47, 16755–16765 Search PubMed.
- Y. Ning, Y.-W. Liu, Y.-S. Meng and J.-L. Zhang, Inorg. Chem., 2018, 57, 1332–1341 Search PubMed.
- R. Herchel, P. Zoufalý and I. Nemec, RSC Adv., 2019, 9, 569–575 Search PubMed.
- D. N. Woolfson, in Advances in Protein Chemistry, ed. D. A. D. Parry and J. M. Squire, Elsevier, Academic Press, 2005, vol. 70, pp. 79–112 Search PubMed.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. C. Bromley, P. J. Booth, R. Leo Brady, A. R. Thomson and D. N. Woolfson, ACS Synth. Biol., 2012, 1, 240–250 Search PubMed.
- D. N. Woolfson, in SubCellular Biochemistry, ed. D. A. D. Parry and J. M. Squire, Springer, Cham, 2017, vol. 82, ch. 2, pp. 35–61 Search PubMed.
- B. Apostolovic, M. Danial and H.-A. Klok, Chem. Soc. Rev., 2010, 39, 3541–3575 Search PubMed.
- C. Nick Pace and J. Martin Scholtz, Biophys. J., 1998, 75, 422–427 Search PubMed.
- Z. N. Mahmoud, S. B. Gunnoo, A. R. Thomson, J. M. Fletcher and D. N. Woolfson, Biomaterials, 2011, 32, 3712–3720 Search PubMed.
- W. D. Kohn, O. D. Monera, C. M. Kay and R. S. Hodges, in Peptides: Chemistry, Structure and Biology. Proceedings of the 14th American Peptide Symposium, ed. P. T. P. Kaumaya and R. S. Hodges, Mayflower Scientific Ltd., Kingswinford, West Midlands, England, 1996, pp. 829–830 Search PubMed.
- W. D. Kohn, C. M. Kay and R. S. Hodges, J. Pept. Res., 1998, 51, 9–18 Search PubMed.
- W. D. Kohn, C. M. Kay, B. D. Sykes and R. S. Hodges, J. Am. Chem. Soc., 1998, 120, 1124–1132 Search PubMed.
- R. Sperling, B. C. Furie, M. Blumenstein, B. Keyt and B. Furie, J. Biol. Chem., 1978, 253, 3898–3906 Search PubMed.
- A. Kashiwada, K. Ishida and K. Matsuda, Bull. Chem. Soc. Jpn., 2007, 80, 2203–2207 Search PubMed.
- M. R. Berwick, D. J. Lewis, A. W. Jones, R. A. Parslow, T. R. Dafforn, H. J. Cooper, J. Wilkie, Z. Pikramenou, M. M. Britton and A. F. A. Peacock, J. Am. Chem. Soc., 2014, 136, 1166–1169 Search PubMed.
- M. R. Berwick, L. N. Slope, C. F. Smith, S. M. King, S. L. Newton, R. B. Gillis, G. G. Adams, A. J. Rowe, S. E. Harding, M. M. Britton and A. F. A. Peacock, Chem. Sci., 2016, 7, 2207–2216 Search PubMed.
- G. De Crescenzo, J. R. Litowski, R. S. Hodges and M. D. O’Connor-McCourt, Biochemistry, 2003, 42, 1754–1763 Search PubMed.
- P. Teare, C. F. Smith, S. J. Adams, S. Anbu, B. Ciani, L. J. C. Jeuken and A. F. A. Peacock, Dalton Trans., 2018, 47, 10784–10790 Search PubMed.
- S. Geremia, L. Di Costanzo, L. Randaccio, D. E. Engel, A. Lombardi, F. Nastri and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 17266–17276 Search PubMed.
- L. Ruckthong, J. A. Stuckey and V. L. Pecoraro, Chem. – Eur. J., 2019, 25, 6773–6787 Search PubMed.
- K.-H. Lee, M. Matzapetakis, S. Mitra, E. N. G. Marsh and V. L. Pecoraro, J. Am. Chem. Soc., 2004, 126, 9178–9179 Search PubMed.
- K.-H. Lee, C. Cabello, L. Hemmingsen, E. N. G. Marsh and V. L. Pecoraro, Angew. Chem., Int. Ed., 2006, 45, 2864–2868 Search PubMed.
- L. Ruckthong, A. Deb, L. Hemmingsen, J. E. Penner-Hahn and V. L. Pecoraro, JBIC, J. Biol. Inorg. Chem., 2018, 23, 123–135 Search PubMed.
- L. N. Slope, M. G. Hill, C. F. Smith, P. Teare, F. J. de Cogan, M. M. Britton and A. F. A. Peacock, Chem. Commun., 2020, 56, 3729–3732 Search PubMed.
- A. Bhagi-Damodaran, P. Hosseinzadeh, E. Mirts, J. Reed, I. D. Petrik and Y. Lu, in Methods in Enzymology, ed. V. L. Pecoraro, Elsevier, Academic Press, 2016, ch. 22, vol. 580, pp. 501–537 Search PubMed.
- J. A. Sigman, H. K. Kim, X. Zhao, J. R. Carey and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3629 LP Search PubMed.
- H. Michel, Biochemistry, 1999, 38, 15129–15140 Search PubMed.
- O. Iranzo, C. Cabello and V. L. Pecoraro, Angew. Chem., Int. Ed., 2007, 46, 6688–6691 Search PubMed.
- M. R. Ghadiri and M. A. Case, Angew. Chem., Int. Ed. Engl., 1993, 32, 1594–1597 Search PubMed.
- G. R. Dieckmann, D. K. McRorie, J. D. Lear, K. A. Sharp, W. F. DeGrado and V. L. Pecoraro, J. Mol. Biol., 1998, 280, 897–912 Search PubMed.
- V. L. Pecoraro, A. F. A. Peacock, O. Iranzo and M. Łuczkowski, in Bioinorganic Chemistry Cellular Systems and Synthetic Models, ed. E. Long and M. J. Baldwin, American Chemical Society, Washington, 2009, vol. 1012, ch. 12, pp. 183–197 Search PubMed.
- S. A. Cotton, Scandium, yttrium and the lanthanides: Applications, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, R. H. Crabtree, C. M. Lukehart, D. A. Attwood and R. A. Scott, 2009 DOI:10.1002/0470862106.ia463.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 Search PubMed.
- E. Potterton, P. Briggs, M. Turkenburg and E. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 1131–1137 Search PubMed.
- S. Takeda, A. Yamashita, K. Maeda and Y. Maéda, Nature, 2003, 424, 35–41 Search PubMed.
- L. Gonzalez, R. A. Brown, D. Richardson and T. Alber, Nat. Struct. Biol., 1996, 3, 1002–1010 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |