
Computational study of silver-catalyzed stereoselective hydroalkylation of alkynes: Pauli repulsion controlled Z/E selectivity†
Lingfei
Hu
a,
Han
Gao
a,
Yanlei
Hu
a,
Xiangying
Lv
 a,
Yan-Bo
Wu
b and
Gang
Lu
*a
a,
Yan-Bo
Wu
b and
Gang
Lu
*a
aSchool of Chemistry and Chemical Engineering, Key Laboratory of Colloid and Interface Chemistry, Ministry of Education, Shandong University, Jinan, Shandong 250100, China. E-mail: ganglu@sdu.edu.cn
bKey Lab for Materials of Energy Conversion and Storage of Shanxi Province and Key Lab of Chemical Biology and Molecular Engineering of Ministry of Education, Institute of Molecular Science, Shanxi University, Taiyuan, Shanxi 030006, China
First published on 26th May 2021
Abstract
The mechanism and origin of stereoselectivity of silver-catalyzed hydroalkylation of alkynes were computationally investigated at the B3LYP-D3BJ/6-311+G(d,p)-SDD//B3LYP/6-31G(d)-LANL2DZ level. The complex of alkynyl trialkylboronate with cationic silver is a key intermediate, which triggers the rate- and stereoselectivity-determining 1,2-migration step. Energy decomposition analysis indicates that the difference of Pauli repulsion dominates the stereoselectivity.
Hydrofunctionalization of terminal alkynes is a straightforward way to produce olefins, commonly used building blocks in organic synthesis. The great challenge is to control the stereochemistry of alkene products. Many efforts in this field contribute to generate E-olefins1 and 1,1-disubstituted olefins.2 The strategy of tuning the formation of thermodynamically less stable Z-olefins remains rare3 (Scheme 1a). In this regard, the Hu group3a reported an iron-catalyzed radical hydroalkylation of aryl acetylenes with varying Z/E selectivity. Recently, the Lalic group3b developed a silver-catalyzed hydroalkylation of terminal alkyl alkynes with extremely high Z selectivity (Scheme 1b). Because efficient approaches for synthesizing Z-olefins are highly desirable, insights into the major factors controlling Z/E selectivity in Ag-catalyzed alkyne hydroalkylations can provide useful guidelines for the development of stereoselective transformations of alkynes.
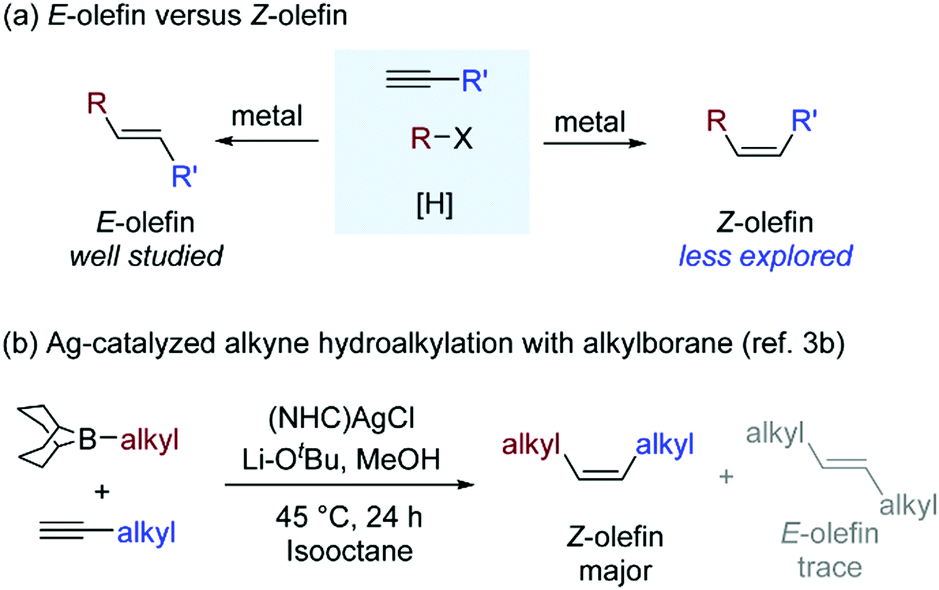 | ||
| Scheme 1 Stereoselective hydroalkylation of terminal alkynes. | ||
Previous experimental4 and computational studies5 on Ag-catalyzed transformations with alkynes revealed that the alkynyl Ag(I) can be easily generated via alkyne C–H activation. After this, the electrophilic boron addition to alkynyl Ag(I) forms the complex of alkynyl trialkylboronate with the η2 Ag coordination to an alkyne π bond (Scheme 2a), which has been experimentally observed.3b Based on the alkynyl trialkylboronate intermediate, the 1,2-migration could occur, which has been utilized in many alkyl, alkenyl/aryl and alkynyl boronate complexes to construct synthetically useful organoboron compounds and derivatives.6–8 However, a more detailed reaction mechanism, including the steps following the 1,2-migration, for the generation of the Z-olefin product is still elusive.
 | ||
| Scheme 2 Possible factors controlling Z/E selectivity. | ||
Furthermore, the 1,2-migration assisted by cationic silver complexes can proceed in a stereoselective manner, i.e., TS-I (anti-to-Ag migration, Scheme 2a) and TS-II (syn-to-Ag migration), followed by protonation and finally affording Z- and E-olefin, respectively. Intuitively, TS-II could be disfavored because the η2 coordination of silver would block the migration from the same side due to steric congestion. Nevertheless, since the σ(B–R) bonding orbital approaches gradually to the alkyne π orbital during the process of R migration from boron to alkyne carbon, a series of possible orbital interactions could also affect the syn/anti selectivity (Scheme 2b). These orbital interactions include not only the stabilizing interactions of σ(B–R) → π*(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) and π(CC) → σ*(B–R), but also the destabilizing interaction between occupied σ(B–R) and π(CC) orbitals. In this work, we identified the dominant factor among those multiple effects for controlling the stereoselectivity by using the distortion/interaction–activation strain model9 and energy decomposition analysis (EDA),10 which have been utilized to successfully explain and predict the origins of reactivity and selectivity in many reactions.11,12
C) and π(CC) → σ*(B–R), but also the destabilizing interaction between occupied σ(B–R) and π(CC) orbitals. In this work, we identified the dominant factor among those multiple effects for controlling the stereoselectivity by using the distortion/interaction–activation strain model9 and energy decomposition analysis (EDA),10 which have been utilized to successfully explain and predict the origins of reactivity and selectivity in many reactions.11,12
We first studied the mechanisms of Ag(I)-catalyzed hydroalkylation of propyne based on DFT calculations at the B3LYP-D3BJ/6-311+G(d,p)-SDD-SMD//B3LYP/6-31G(d)-LANL2DZ level of theory, which shows consistent results with other methods (see computational details in the ESI†). A series of Ag(I) complexes, such as L-Ag-Cl, L–Ag–OMe, and L-Ag–OtBu were considered under the experimental conditions. The alkyne C–H activations by all these species proceed smoothly (see details in Fig. S1, ESI†). The propyne C–H activation by the complex of L-Ag–OMe with LiOtBu (2-TS) is shown in Fig. 1. This leads to the formation of alkynyl Ag(I) intermediate 4.
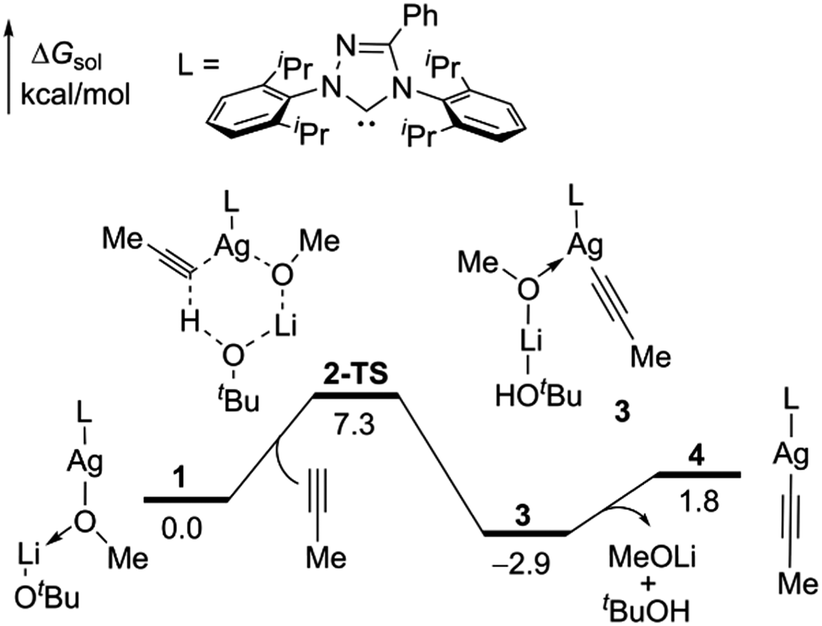 | ||
| Fig. 1 Energy profile of the formation of alkynyl Ag(I) species. | ||
Trialkylboron 5 can add to alkynyl Ag(I) 4 with a low barrier (6-TS, Fig. 2), which generates alkynyl boronate intermediate 7 with Ag coordination in a η2 fashion. After this, the syn/anti-to-Ag 1,2-migrations (8-TS and 9-TS, Fig. 2 and 3) occur and form alkenyl Ag intermediates (10 and 11), respectively. In the overall energy profile shown in Fig. 1, 2 and 4, the 1,2-migration step has the highest barrier (ΔG‡3→8-TS = 24.7 kcal mol−1) and is an irreversible process (10 → 12-TS having a lower barrier than 10 → 8-TS). Thus, the 1,2-migration determines both the reactivity and stereoselectivity in Ag-catalyzed propyne hydroalkylation (see below for detailed discussions on the origin of stereochemistry).
 | ||
| Fig. 2 Energy profiles of competing 1,2-alkyl migrations. | ||
 | ||
| Fig. 3 Optimized geometries of 1,2-migration transition states. | ||
 | ||
| Fig. 4 Energy profiles of the Ag–C bond protonation and transmetalation to generate a Z-olefin product. | ||
Fig. 4 shows the subsequent transformations of alkenyl Ag intermediate 10. Generally, protonations of the Ag–C and C–B bonds in 10 are required to deliver the major Z-olefin product. Our results indicate that the protonation of the Ag–C bond (12-TS) is much more favorable than that of the B–C bond (12a-TS) in the presence of MeOH. In addition, the Ag–C protonation is significantly promoted by the coordination of MeOH with a boron moiety (12-TSvs.12b-TS). Since the B–C bond is difficult to be protonated, a transmetalation step can easily change the B–C(alkenyl) bond to a new Ag–C(alkenyl) bond via14-TS with a barrier of 12.5 kcal mol−1. Then, the second Ag–C bond protonation (16-TS, ΔG‡ = 12.6 kcal mol−1) of the formed alkenyl Ag(I) (15) can finally deliver the Z-olefin product. This step is also assisted by the boron coordination to MeOH.
Taken together, the most favorable pathway for Z-olefin includes alkyne C–H activation (Fig. 1), anti-1,2-migration (Fig. 2), the first Ag–C bond protonation, transmetalation and the second Ag–C bond protonation (Fig. 4). Other less favorable competing pathways are given in Fig. S2 and S3 (ESI†).
Next, we investigate the origin of syn/anti selectivity of 1,2-migration. The anti-migration (8-TS) is 2.8 kcal mol−1 lower than the syn-migration (9-TS), which agrees with the experimentally observed stereoselectivity for Z-olefin.3b To understand the origin of this barrier difference, we performed EDA calculations at the B3LYP-D3BJ/6-311+G(d,p)-LANL2DZ level. The transition states were first separated into two reactive fragments: the L-Ag-alkynyl fragment and the boron fragment (highlighted in yellow in Fig. 3). Then, the activation energies (ΔE‡) of 8-TS and 9-TS were dissected into six energy terms, including distortion (ΔEdist), Pauli repulsion (ΔEPauli), electrostatics (ΔEelstat), polarization (ΔEpol), charge transfer (ΔEct) and dispersion (ΔEdisp), along the intrinsic reaction coordinates (IRC) to figure out the major factors affecting the stereochemistry.
As shown in Fig. 5a, among the six energy terms, only the distortion (ΔEdist) and Pauli repulsion (ΔEPauli) energies positively contribute to the lower barrier of 8-TS than 9-TS along their reaction coordinates. Although other types of interactions favor 9-TS, they are surpassed by the sum of ΔEdist and ΔEPauli (see details in Tables S3 and S4, ESI†). The results reveal that the preference of anti-migration (8-TS) is because of having weaker destabilizing interactions, not due to stronger stabilizing interactions.
 | ||
| Fig. 5 (a) EDA results along IRC for the two stereoselective transition states (8-TS and 9-TS). (b) Origin of the difference of Pauli repulsion. (c) Orbital interactions between the cationic silver and alkynyl boronate fragments in complex 7. | ||
Clearly, the difference of Pauli repulsion is the most significant effect on destabilizing 9-TS (Fig. 5a). The deformation of two fragments also destabilizes 9-TS albeit with relatively low importance. The effect of distortion on the syn/anti selectivity is consistent with the chemical intuition that the alkyl group migrated from the same side of Ag is rejected by the η2-coordinated Ag complex, thus showing a relatively large distorted energy. However, this effect is less significant. That is because the Ag complex shifts to the Me-substituted alkyne carbon in both 8-TS and 9-TS, and the change of η2 → η1 coordination leads to comparable deformations with respect to the complex 7. Furthermore, the migrated alkyl group repels the Me and L-Ag moieties in 8-TS and 9-TS, respectively. The similar changes in the bond angles of ∠AgC2C3 (114° vs. 129°) and ∠C1C2C3 (128° vs. 116°) also result in comparable deformations (Fig. 3).
To understand the dominant role of Pauli repulsion in differentiating 8-TS and 9-TS, we focus on the two-orbital-four-electron interaction (i.e., closed shell repulsion) between the filled B–C σ orbital and alkyne in-plane π orbital. Fig. 5b shows these bonding orbitals of the boron and L-Ag-alkyne fragments. Clearly, the electron of the alkyne in-plane π orbital is redistributed, accumulating at the same side of Ag and depleting at the opposite side. The complementary occupied-virtual pairs (COVPs) were used to analyze this rearrangment of the π orbital based on the complex 7. The COVPs results in Fig. 5c indicate that the charge transfer from the alkyne in-plane π orbital to the empty Ag s orbital, much greater than the reverse electron transfer from the Ag d orbital to the alkyne π* antibonding orbital, is the most significant donor–acceptor interaction for the change of alkyne π electron density. This is also supported by the charge density difference plot (see details in Fig. S4, ESI†). Therefore, the Pr migration suffers from stronger σ)(π Pauli repulsion when approaching from the Ag side, and weaker σ)(π Pauli repulsion from the Me side. The difference of Pauli repulsion is significant enough to destabilize 9-TS, thus favoring the anti-migration (8-TS).
In summary, we present a computational study on the mechanism and origin of stereoselectivity in Ag-catalyzed hydroalkylation of terminal alkynes with boron compounds. The irreversible 1,2-migration is the key step, which determines both the reactivity and stereoselectivity. By the use of energy decomposition analysis, we found that the σ)(π Pauli repulsion is the dominant factor for destabilizing the syn-1,2-migration, thus favoring the anti-1,2-migration pathway. The difference of Pauli repulsion between the syn- and anti-migration is attributed to the cationic Ag-induced unequal redistribution of the in-plane π orbital of alkyne fragments, thus exerting different σ)(π Pauli repulsion when the B–C σ bonding orbital approaches from above and below. We anticipate that the chemically meaningful insights of manipulating destabilizing Pauli repulsion (closed shell repulsion) can be potentially applicable to improve the selectivity of reactions with various boronate complexes.
This work was supported by the National Natural Science Foundation of China (No. 21973055), the Natural Science Foundation of Shandong Province (ZR2019MB049), the Taishan Scholar of Shandong Province (No. tsqn201812013) and the Qilu Young Scholar of Shandong University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. R. Uehling, A. M. Suess and G. Lalic, J. Am. Chem. Soc., 2015, 137, 1424–1427 CrossRef CAS PubMed; (b) M. K. Armstrong, M. B. Goodstein and G. Lalic, J. Am. Chem. Soc., 2018, 140, 10233–10241 CrossRef CAS PubMed; (c) A. Hazra, J. Chen and G. Lalic, J. Am. Chem. Soc., 2019, 141, 12464–12469 CrossRef CAS PubMed; (d) L. Yu, L. Lv, Z. Qiu, Z. Chen, Z. Tan, Y.-F. Liang and C.-J. Li, Angew. Chem., Int. Ed., 2020, 59, 14009–14013 CrossRef CAS PubMed; (e) K. Nakamura and T. Nishikata, ACS Catal., 2017, 7, 1049–1052 CrossRef CAS; (f) F. Ye, J. Chen and T. Ritter, J. Am. Chem. Soc., 2017, 139, 7184–7187 CrossRef CAS PubMed.
- (a) X.-Y. Lu, J.-H. Liu, X. Lu, Z.-Q. Zhang, T.-J. Gong, B. Xiao and Y. Fu, Chem. Commun., 2016, 52, 5324–5327 RSC; (b) A. Sabarre and J. Love, Org. Lett., 2008, 10, 3941–3944 CrossRef CAS PubMed; (c) J. R. DeBergh, K. M. Spivey and J. M. Ready, J. Am. Chem. Soc., 2008, 130, 7828–7829 CrossRef CAS PubMed; (d) T. Li, Y. Yang, B. Luo, B. Li, L. Zong, W. Kong, H. Yang, X. Cheng and L. Zhang, Org. Lett., 2020, 22, 6045–6049 CrossRef CAS PubMed; (e) X. Zhou, Y. Luo, L. Kong, Y. Xu, G. Zheng, Y. Lan and X. Li, ACS Catal., 2017, 7, 7296–7304 CrossRef CAS.
- (a) C. W. Cheung, F. E. Zhurkin and X. Hu, J. Am. Chem. Soc., 2015, 17, 4932–4935 CrossRef PubMed; (b) M. T. Lee, M. B. Goodstein and G. Lalic, J. Am. Chem. Soc., 2019, 141, 17086–17091 CrossRef CAS PubMed.
- (a) P. Sivaguru, S. Cao, K. R. Babu and X. Bi, Acc. Chem. Res., 2020, 5, 662–675 CrossRef PubMed; (b) M. Li, W. Wu and H. Jiang, ChemCatChem, 2020, 12, 5034–5050 CrossRef CAS.
- (a) J. Jover and F. Maseras, J. Org. Chem., 2014, 79, 11981–11987 CrossRef CAS PubMed; (b) C. Liu, Y. Luo, W. Zhang, J. Qu and X. Lu, Organometallics, 2014, 33, 2984–2989 CrossRef CAS; (c) S. Cao, Q. Ji, H. Li, M. Pang, H. Yuan, J. Zhang and X. Bi, J. Am. Chem. Soc., 2020, 142, 7083–7091 CrossRef CAS PubMed.
- (a) A. Fawcett, T. Biberger and V. K. Aggarwal, Nat. Chem., 2019, 11, 117–122 CrossRef CAS PubMed; (b) A. Fawcett, A. Murtaza, C. H. U. Gregson and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 4573–4578 CrossRef CAS PubMed; (c) M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511–9515 CrossRef CAS PubMed; (d) S. H. Bennett, A. Fawcett, E. H. Denton, T. Biberger, V. Fasano, N. Winter and V. K. Aggarwal, J. Am. Chem. Soc., 2020, 142, 16766–16775 CrossRef CAS PubMed.
- (a) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70 CrossRef CAS PubMed; (b) M. Chierchia, C. Law and J. P. Morken, Angew. Chem., Int. Ed., 2017, 56, 11870–11874 CrossRef CAS PubMed; (c) J. A. Myhill, C. A. Wilhelmsen, L. Zhang and J. P. Morken, J. Am. Chem. Soc., 2018, 140, 15181–15185 CrossRef CAS PubMed; (d) Z. He, F. Song, H. Sun and Y. Huang, J. Am. Chem. Soc., 2018, 140, 2693–2699 CrossRef CAS PubMed.
- (a) A. Pelter, K. J. Gould and L. A. P. Kane-Maguire, J. Chem. Soc., Chem. Commun., 1974, 1029–1030 RSC; (b) B. Wrackmeyer, Coord. Chem. Rev., 1995, 145, 125–156 CAS.
- (a) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed; (b) I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC; (c) P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Nat. Protoc., 2020, 15, 649–667 CrossRef CAS PubMed.
- (a) K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS; (b) R. Z. Khaliullin, E. A. Cobar, R. C. Lochan, A. T. Bell and M. Head-Gordon, J. Phys. Chem. A, 2007, 111, 8753–8765 CrossRef CAS PubMed; (c) M. V. Hopffgarten and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 43–62 Search PubMed; (d) L. Zhao, M. Hermann, W. H. E. Schwarz and G. Frenking, Nat. Rev. Chem., 2019, 3, 48–63 CrossRef CAS; (e) L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1345 Search PubMed.
- (a) P. Vermeeren, T. A. Hamlin, I. Fernández and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2020, 59, 6201–6206 CrossRef CAS PubMed; (b) T. A. Hamlin, I. Fernández and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2019, 58, 8922–8926 CrossRef CAS PubMed; (c) P. Vermeeren, T. A. Hamlin, I. Fernández and F. M. Bickelhaupt, Chem. Sci., 2020, 11, 8105–8112 RSC; (d) T. A. Hamlin, F. M. Bickelhaupt and I. Fernández, Acc. Chem. Res., 2021, 54, 1972–1981 CrossRef CAS PubMed.
- (a) X. Qi, D. G. Kohler, K. L. Hull and P. Liu, J. Am. Chem. Soc., 2019, 141, 11892–11904 CrossRef CAS PubMed; (b) A. A. Thomas, K. Speck, I. Kevlishvili, Z. Lu, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 13976–13984 CrossRef CAS PubMed; (c) N. I. Saper, A. Ohgi, D. W. Small, K. Semba, Y. Nakao and J. F. Hartwig, Nat. Chem., 2020, 12, 276–283 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, additional discussions and Cartesian coordinates and energies of computed structures. See DOI: 10.1039/d1cc01917a |
| This journal is © The Royal Society of Chemistry 2021 |