
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Borane catalysed cyclopropenation of arylacetylenes†
Katarina
Stefkova
 ,
Matthew J.
Heard
,
Ayan
Dasgupta
* and
Rebecca L.
Melen
*
,
Matthew J.
Heard
,
Ayan
Dasgupta
* and
Rebecca L.
Melen
*
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, Cymru/Wales, UK. E-mail: DasguptaA3@cardiff.ac.uk; MelenR@cardiff.ac.uk
First published on 8th June 2021
Abstract
Triarylboranes have gained substantial attention as catalysts for C–C bond forming reactions due to their remarkable catalytic activities. Herein, we report B(C6F5)3 catalysed cyclopropenation of a wide variety of arylacetylenes using donor–acceptor diazoesters. A mild reaction protocol has been developed for the synthesis of functionalised cyclopropenes (33 examples) in good to excellent yields.
Transition metal catalysed C–C bond forming reactions overwhelm the chemical literature.1 Although the use of precious transition metal catalysts has achieved immense success, metal impurities in the final compounds are often unavoidable. This is particularly significant when considering products taken into the body where toxic metal contamination must be kept to a minimum. Over the last few years, main group-based catalysts have been extensively investigated as substantial alternative to the precious transition metals.2 More precisely, the Lewis acidic triarylboranes3 have found multitude applications towards C–C bond forming reactions.4,5 The presence of empty d-orbitals in transition metals allows them to lend or remove electrons from the coupling partners, and thus can be employed as a catalyst for wide variety of reactions.6 Likewise, the empty p-orbital of the central boron atom of Lewis acids renders them strongly electrophilic in nature and therefore they can readily react with Lewis bases by accepting a lone pair of electrons.7 Relating to this, an important initial contribution made by Zhang in 2016,8 showed that B(C6F5)3 could act as a catalyst for the ortho-selective C–H alkylation of unprotected phenols with α-aryl α-diazoesters. The mechanism for this reaction was revealed computationally to be the activation of the diazoester through O → B adduct formation to generate carbenes.9 Therefore, by using diazoester precursors, a carbene transfer reactions can be carried out using B(C6F5)3 as a catalyst.10
Carbene transfer reactions are one of the most fundamental reactions in organic synthesis and widespread studies have been conducted to explore the synthetic utility of carbenes for making a variety of novel compounds.11 Recently, we12 and Wilkerson-Hill13 observed that catalytic amounts of B(C6F5)3 enable the cyclopropanation reactions of styrenes (Scheme 1A) using α-aryl α-diazoesters. This exciting outcome motivated us to investigate this reactivity further to see if arylacetylenes could also be used as substrates in reactions with α-aryl α-diazoesters using B(C6F5)3 as a catalyst. This reaction, cyclopropenation, has been largely investigated using precious transition metals, such as Rh,14 Ir,15 Ag,16 and Au.17 Nonetheless, the use of non-precious transition metals, including Cu18 and Co19 (Scheme 1B), have also been reported. Typically, in the presence of a metal catalyst, diazoesters afford a metal-carbenoid species which then readily undergoes a [2+1] cycloaddition with an arylacetylene to form the 3-membered carbocycle. Recently, Koenigs et al. revealed that cyclopropenation of arylacetylenes using α-aryl α-diazoesters is also possible by employing visible light (blue light; 470 nm).20
 | ||
| Scheme 1 General approach for cyclopropenation/cyclopropanation reaction. | ||
We initiated our studies into the cyclopropenation reaction using phenylacetylene (1.3 equiv.) and α-aryl α-diazoester 1a (1 equiv.) as model substrates (Table 1). The reaction between 1a and phenylacetylene showed no formation of the cyclopropene product (3i) in absence of any catalyst at both ambient temperature and at reflux in CH2Cl2 (Table 1, entries 1 and 2). Addition of BF3·OEt2 as a Lewis acid catalyst (20 mol%) also showed no formation of the desired carbocycle (3i) and only decomposition of the diazo compound into the homocoupled product was observed (Table 1, entry 3). 40 mol% of the Brønsted acid (TfOH, triflic acid) also failed to promote the reaction (Table 1, entry 4). When 20 mol% B(C6F5)3 was employed for the reaction, no product formation was observed at ambient temperature, however reaction at 45 °C afforded 3i in 48% yield after 24 h (Table 1, entries 5 and 6). Switching the solvent from CH2Cl2 to C2H4Cl2 slightly improved the yield of 3i to 57% but still did not give satisfactory results (Table 1, entry 7). Increasing the reaction temperature further to 70 °C however was detrimental to the reaction leading to the formation of a complicated reaction mixture and the isolation of the desired product 3i failed (Table 1, entry 8). Reducing the catalytic loading of B(C6F5)3 from 20 mol% to 10 mol% showed improvement in the yield of 3i to 65%. However, reducing catalytic loadings further to 5 mol% gave lower yields of the desired carbocycle 3i of 32% (Table 1, entries 9 and 10). Additionally, we tested other triarylfluoroborane catalysts for the cyclopropenation reaction and we observed that although 10 mol% (2,4,6-F3C6H2)3B [(2,4,6-ArF)3B] afforded 3i in poor yields of 28%, the Lewis acidic boranes (3,4,5-F3C6H2)3B [(3,4,5-ArF)3B] and (3,5-CF3C6H3)3B [(3,5-CF3-ArF)3B] completely failed to catalyse the reaction, and no product formation was detected (Table 1, entries 11–13).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Solvent | Temp. (°C) | Time (h) | Yielda (%) |
| All the reactions were carried out on a 0.1 mmol scale. 1a (1 equiv.), phenylacetylene (1.3 equiv.), and 1.5 mL solvent.a Reported yields are isolated.b Phenylacetylene (1 equiv.), 1a (1.3 equiv.), and 1.5 mL of solvent. | ||||||
| 1 | None | — | CH2Cl2 | r.t. | 24 | — |
| 2 | None | — | CH2Cl2 | 45 | 24 | — |
| 3 | BF3·OEt2 | 20 | CH2Cl2 | 45 | 24 | — |
| 4 | TfOH | 40 | CH2Cl2 | 45 | 24 | — |
| 5 | B(C6F5)3 | 20 | CH2Cl2 | r.t. | 24 | — |
| 6 | B(C6F5)3 | 20 | CH2Cl2 | 45 | 24 | 48 |
| 7 | B(C6F5)3 | 20 | C2H4Cl2 | 50 | 22 | 57 |
| 8 | B(C6F5)3 | 20 | C2H4Cl2 | 70 | 18 | — |
| 9 | B(C6F5)3 | 10 | C2H4Cl2 | 50 | 24 | 65 |
| 10 | B(C6F5)3 | 5 | C2H4Cl2 | 50 | 28 | 32 |
| 11 | (2,4,6-ArF)3B | 10 | C2H4Cl2 | 50 | 24 | 28 |
| 12 | (3,4,5-ArF)3B | 10 | C2H4Cl2 | 40 | 24 | — |
| 13 | (3,5-CF3-ArF)3B | 10 | C2H4Cl2 | 50 | 24 | — |
| 14 | B(C6F5)3b | 10 | C2H4Cl2 | 50 | 24 | 75 |
Interestingly, the yield of 3i was further improved to 75% when we used a slight excess of 1a (1.3 equiv.) (Table 1, entry 14). Thus our optimised reaction conditions were a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3 stoichiometric ratio of phenylacetylene:1a and carrying out the reaction in C2H4Cl2 at 50 °C using 10 mol% B(C6F5)3.
:1.3 stoichiometric ratio of phenylacetylene:1a and carrying out the reaction in C2H4Cl2 at 50 °C using 10 mol% B(C6F5)3.
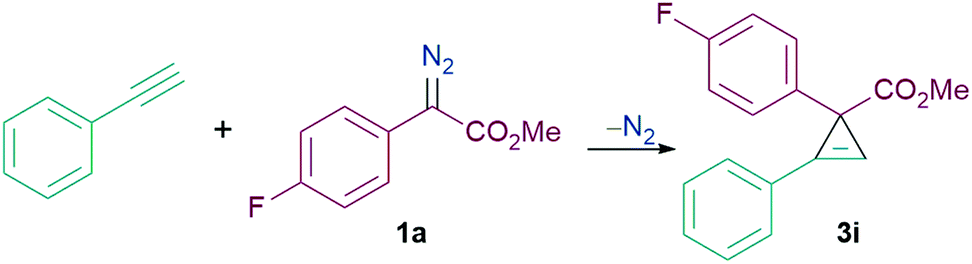
With the optimised conditions in hand, we then explored the scope of the reaction with various α-aryl α-diazoesters (1) and arylacetylenes (Scheme 2). Firstly, terminal arylacetylenes were explored bearing electron-withdrawing, neutral, and electron releasing groups, for the cyclopropenation reaction with different α-aryl α-diazoesters (1a–g) to generate the products 3a–3ae in 30–84% yields. Of these, products 3n and 3o could be recrystallised by vapour diffusion from CH2Cl2/pentane and their structures elucidated by single crystal X-ray diffraction (Fig. 1). Lower yields were observed with strongly electron withdrawing (p-CF3) or electron releasing (p-OMe, p-Me) arylacetylenes giving 3a,b, u–w in less than 50% yield. Another observation was that the p-F or o-F substituted α-aryl α-diazoesters (1a and 1b respectively) gave better yields than the p-OMe substituted α-aryl α-diazoester (1g) when combined with the same arylacetylene. Interestingly, the vinyl diazoacetate (methyl (E)-2-diazo-4-phenylbut-3-enoate, (1h), symmetrical diisopropyl 2-diazomalonate (1i), diazodimedone (1j), ethyl 2-diazoacetate (1k), and methyl 2-diazo-2-(3-fluorophenyl)acetate (1l) failed to react with arylacetylenes to afford the desired carbocycles. Aliphatic terminal acetylenes including tert-butylacetylene and hex-1-yne, as well as ethynyltrimethylsilane were also unsuccessful for the reaction.
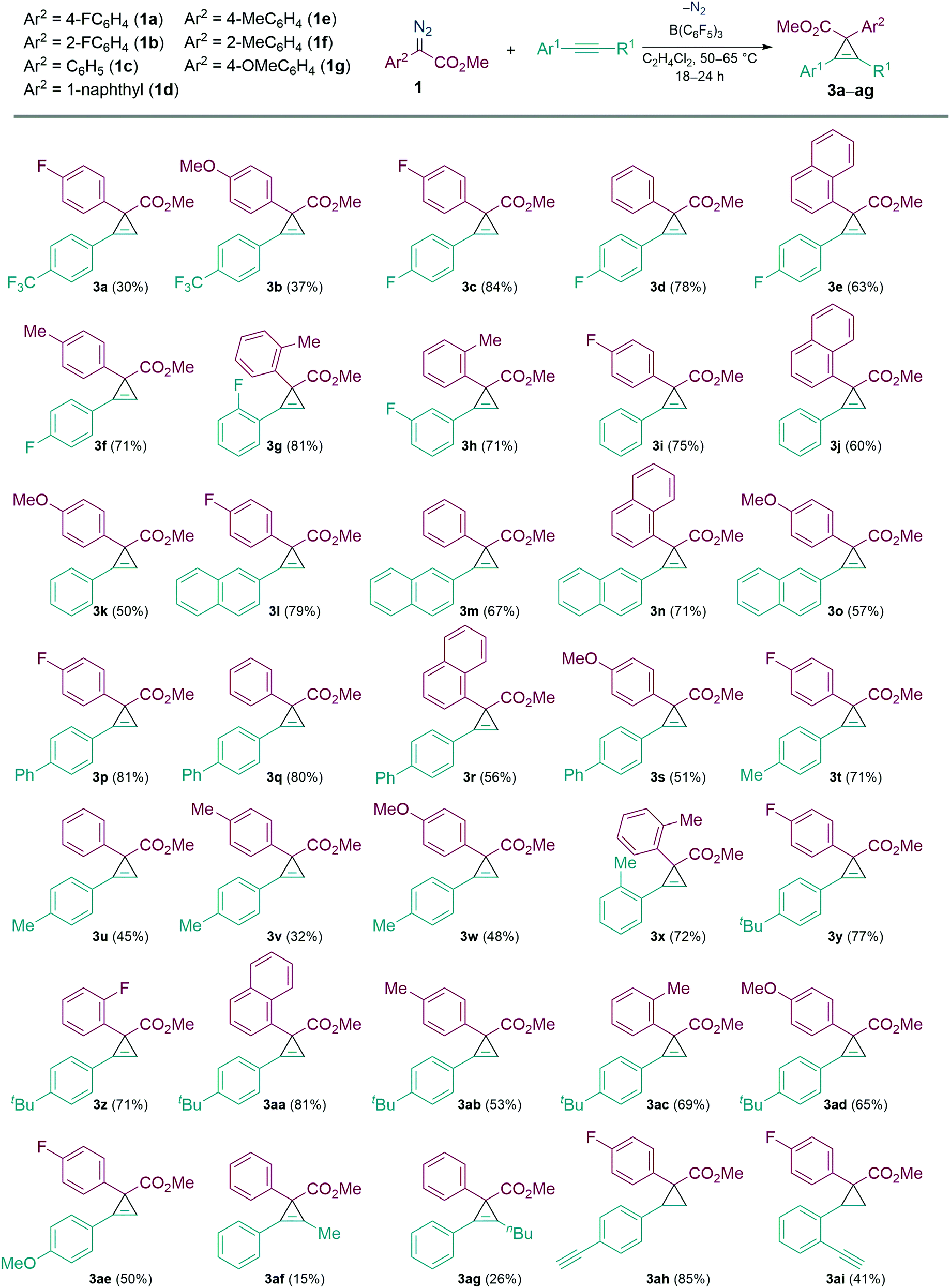 | ||
| Scheme 2 Substrate scope for the cyclopropenation of arylacetylenes. Yields reported are isolated. All the reactions were carried out in 0.1 mmol scale, 10 mol% B(C6F5)3 was used. 1 (1.3 equiv.), phenylacetylene (1 equiv.), and 1.5 mL of solvents were used. | ||
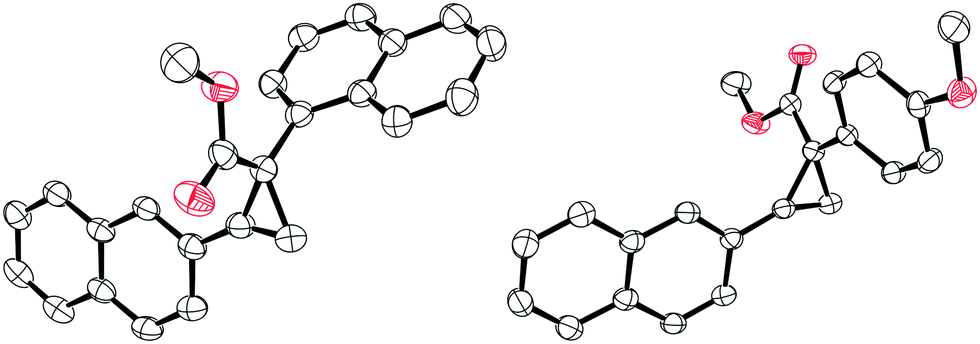 | ||
| Fig. 1 Solid-state structure of compound 3n (left) and 3o (right). Thermal ellipsoids drawn at 50% probability. Carbon: black; oxygen: red. H atoms omitted for clarity. | ||
We then examined the reactivities of the internal alkynes in the cyclopropenation reaction with limited success. However, when 1-phenyl-1-propyne and hex-1-ynylbenzene (2a) were reacted with 1c, the desired carbocycles 3af and 3ag were isolated in poor yields of 15% and 26% respectively. Other internal alkynes such as 1,2-diphenylethyne, trimethyl(phenylethynyl)silane, and 1-(prop-1-yn-1-yl)-4-(trifluoromethyl)benzene failed to react completely.
Following this we investigated the selectivity of the carbocycle formation (cyclopropanation versus cyclopropenation) when both alkene and alkyne functionalities are present. For this competition study, the intramolecular alkene/alkyne compounds 1-ethynyl-4-vinylbenzene (2b) and 1-ethynyl-2-vinylbenzene (2c) were tested in the B(C6F5)3 catalysed reaction with 1a. In both cases cyclopropanation was favoured over cyclopropenation giving the cyclopropane carbocycle as a single diastereoisomer in 88% (3ah) and 41% (3ai) yields, respectively.
Unfortunately, attempts to synthesise 3-membered heterocycles from the insertion of the carbene into C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN or C
O, CN or C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds failed. Using the optimised reaction conditions, the reaction between benzaldehyde and 1c was examined with the goal to produce the corresponding epoxide. However, multinuclear spectroscopic data of the isolated compound confirmed the formation of methyl 3-oxo-2,3-diphenylpropanoate (see ESI†) formed from the homologation of benzaldehyde with the diazo compound (Roskamp–Feng reaction).21
N bonds failed. Using the optimised reaction conditions, the reaction between benzaldehyde and 1c was examined with the goal to produce the corresponding epoxide. However, multinuclear spectroscopic data of the isolated compound confirmed the formation of methyl 3-oxo-2,3-diphenylpropanoate (see ESI†) formed from the homologation of benzaldehyde with the diazo compound (Roskamp–Feng reaction).21
We propose the mechanism of the cyclopropenation reaction to proceed in a similar manner to that for the cyclopropanation reaction reported in our previous studies12 (Fig. 2). Initially, the Lewis acidic B(C6F5)3 binds effectively with the ester functionality of the α-aryl α-diazoester 1. This facilitates loss of N2 forming the highly electron deficient intermediate I and its resonance form I′. Subsequently the reactive carbene intermediate can then react with the nucleophilic arylacetylene forming intermediate II. The generation of the carbocationic centre in II explains the need for arylacetylenes in the reaction to stabilise this intermediate. Finally, attack of the boron enolate onto the carbocation in II then generates the product and regenerates the catalyst.
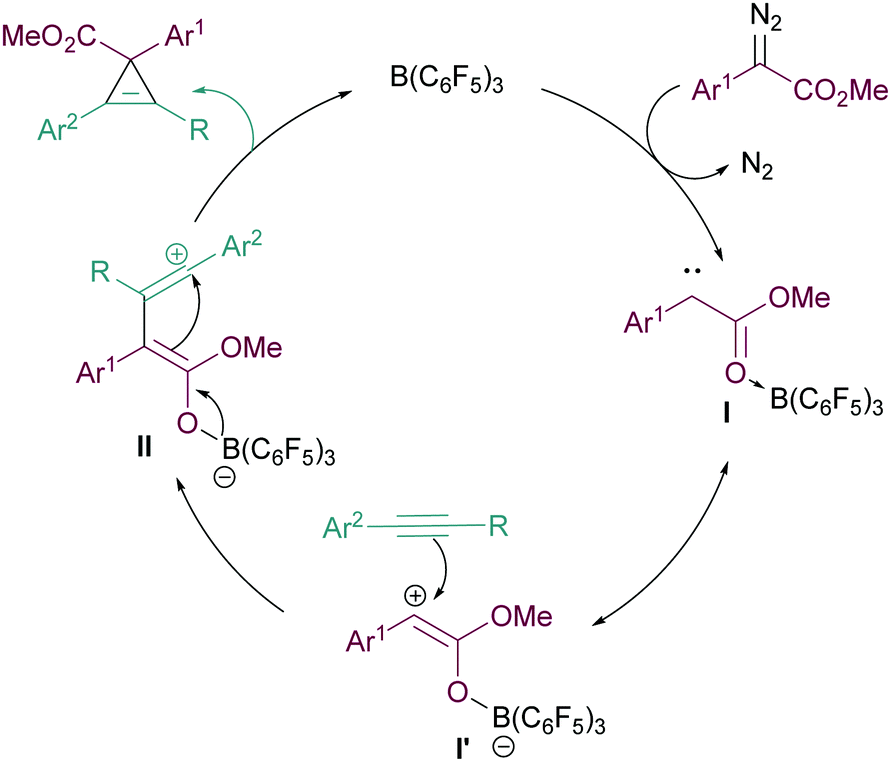 | ||
| Fig. 2 Proposed reaction mechanism for the cyclopropenation reaction. | ||
In conclusion, a metal-free mild reaction protocol has been developed for the cyclopropenation of alkynes using diazo compounds. Our studies demonstrate that catalytic amounts of B(C6F5)3 readily react with α-aryl α-diazoesters to promote the carbene transfer reaction when reacted with arylacetylenes generating the desired 3-membered carbocycle. A wide range of substrates were investigated and good to excellent yields of the cyclopropenated products were obtained. This methodology adds to the ever-increasing range of reactions that the Lewis acid B(C6F5)3 can catalyse.
AD and RLM are grateful to the EPSRC for funding and the awarding of an EPSRC Early Career Fellowship (EP/R026912/1). Information about the data that underpins the results presented in this article, including how to access them, can be found in the Cardiff University data catalogue at http://doi.org/10.17035/d.2021.0136187455.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- For selected reviews see: (a) J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 Search PubMed; (b) S. G. Modha, V. P. Mehta and E. V. der Eycken, Chem. Soc. Rev., 2013, 42, 5042–5055 Search PubMed.
- For selected articles see: (a) J. R. Ludwig and C. S. Schindler, Chemistry, 2017, 2, 313–316 Search PubMed; (b) Q.-L. Zhou, Angew. Chem., Int. Ed., 2016, 55, 5352–5353 Search PubMed.
- S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279–4285 Search PubMed.
- For selected reviews see: (a) J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 Search PubMed; (b) L. Deloux and M. Srebnik, Chem. Rev., 1993, 93, 763–784 Search PubMed.
- For selected reviews see: (a) G. Kumar, S. Roy and I. Chatterjee, Org. Biomol. Chem., 2021, 19, 1230–1267 Search PubMed; (b) Y. Ma, S.-J. Lou and Z. Hou, Chem. Soc. Rev., 2021, 50, 1945–1967 Search PubMed.
- J. Takaya, Chem. Sci., 2021, 12, 1964–1981 Search PubMed.
- R. L. Melen, Science, 2019, 363, 479–484 Search PubMed.
- Z. Yu, Y. Li, J. Shi, B. Ma, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 14807–14811 Search PubMed.
- Q. Zhang, X.-F. Zhang, M. Li, C. Li, J.-Q. Liu, Y.-Y. Jiang, X. Ji, L. Liu and Y.-C. Wu, J. Org. Chem., 2019, 84, 14508–14519 Search PubMed.
- For selected article see: (a) S. Rao, R. Kapanaiah and K. R. Prabhu, Adv. Synth. Catal., 2019, 361, 1301–1306 Search PubMed; (b) H. H. San, C.-Y. Wang, H.-P. Zeng, S.-T. Fu, M. Jiang and X.-Y. Tang, J. Org. Chem., 2019, 84, 4478–4485 Search PubMed; (c) H. H. San, S.-J. Wang, M. Jiang and X.-Y. Tang, Org. Lett., 2018, 20, 4672–4676 Search PubMed.
- For selected reviews see: (a) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 Search PubMed; (b) L.-W. Ye, X.-Q. Zhu, R. L. Sahani, Y. Xu, P.-C. Qian and R.-S. Liu, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00348; (c) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 Search PubMed; (d) L. Liu and J. Zhang, Chem. Soc. Rev., 2016, 45, 506–516 Search PubMed; (e) S.-F. Zhu and Q.-L. Zhou, Natl. Sci. Rev., 2014, 1, 580–603 Search PubMed.
- A. Dasgupta, R. Babaahmadi, B. Slater, B. F. Yates, A. Ariafard and R. L. Melen, Chemistry, 2020, 6, 2364–2381 Search PubMed.
- J. P. Mancinelli and S. M. Wilkerson-Hill, ACS Catal., 2020, 10, 11171–11176 Search PubMed.
- For a selected article see: J. F. Briones, J. Hansen, K. I. Hardcastle, J. Autschbach and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 17211–17215 Search PubMed.
- M. Uehara, H. Suematsu, Y. Yasutomi and T. Katsuki, J. Am. Chem. Soc., 2010, 133, 170–171 Search PubMed.
- J. F. Briones and H. M. L. Davies, Org. Lett., 2011, 13, 3984–3987 Search PubMed.
- J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 11916–11919 Search PubMed.
- M. M. Díaz-Requejo, M. A. Mairena, T. R. Belderrain, M. C. Nicasio, S. Trofimenko and P. J. Pérez, Chem. Commun., 2001, 1804–1805 Search PubMed.
- X. Cui, X. Xu, H. Lu, S. Zhu, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 3304–3307 Search PubMed.
- R. Hommelsheim, Y. Guo, Z. Yang, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2019, 58, 1203–1207 Search PubMed.
- W. Li, J. Wang, X. Hu, K. Shen, W. Wang, Y. Chu, L. Lin, X. Liu and X. Feng, J. Am. Chem. Soc., 2010, 132, 8532–8533 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR data, X-ray data. CCDC 2072872 and 2072873. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc01856f |
| This journal is © The Royal Society of Chemistry 2021 |