
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formaldehyde regulates tetrahydrofolate stability and thymidylate synthase catalysis†
Xiaolei
Chen
 a,
Sara Y.
Chothia
a,
Jaswir
Basran
b and
Richard J.
Hopkinson
*a
a,
Sara Y.
Chothia
a,
Jaswir
Basran
b and
Richard J.
Hopkinson
*a
aLeicester Institute of Structural and Chemical Biology and School of Chemistry, University of Leicester, Henry Wellcome Building, Lancaster Road, Leicester, LE1 7RH, UK. E-mail: richard.hopkinson@leicester.ac.uk
bDepartment of Molecular and Cell Biology, University of Leicester, Henry Wellcome Building, Lancaster Road, Leicester, LE1 7RH, UK
First published on 17th May 2021
Abstract
Tetrahydrofolic acid and formaldehyde are key human metabolites but their physiologically relevant chemistry is undefined. Our NMR studies confirm formaldehyde as a product of tetrahydrofolic acid degradation but also reveal their reaction regulates the stability of tetrahydrofolic acid. These observations identify a novel non-enzymatic feedback mechanism regulating formaldehyde and folate metabolism that has important implications for folate-targeting chemotherapy in cancer and other diseases.
The folate pathway is a ubiquitous metabolic pathway that mediates one-carbon transfer processes. While the folate pathway involves multiple substrates and products, its key metabolic unit is tetrahydrofolic acid (THF), which is produced in humans via reduction of folic acid (vitamin B9, Fig. 1).1 Depletion of the folate pathway is toxic and correlates with increased risk of disease and developmental defects.2–4 Consequently, folic acid is a common dietary additive,5 while antagonistic folic acid derivatives are used as antimicrobial and cancer chemotherapies.6,7
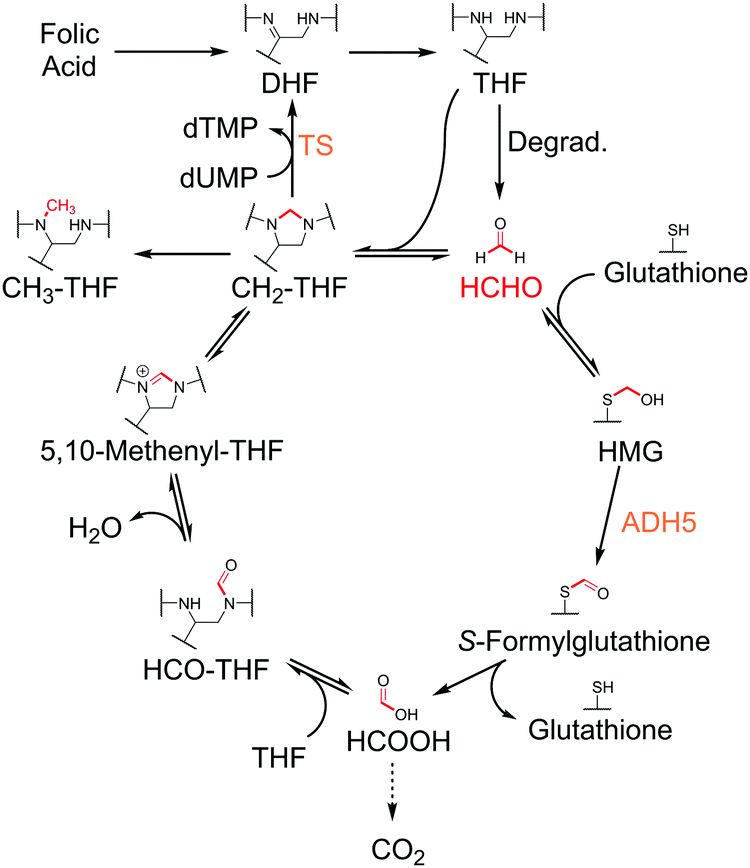 | ||
| Fig. 1 Mechanisms of folic acid- and glutathione-mediated HCHO metabolism. HCHO is formed in cells by multiple mechanisms, including by the degradation of THF. THF also reacts with HCHO to form CH2-THF, which provides the methyl group for TS-catalysed methylation of dUMP. CH2-THF can also be enzymatically converted to other THF derivatives such as 5,10-methenyl-THF, 5-methyl-THF (CH3-THF) and 10-formyl-THF (HCO-THF). HCHO also reacts with glutathione to form S-hydroxymethylglutathione (HMG), which undergoes ADH5-catalysed oxidation to form S-formylglutathione. Subsequent hydrolysis produces formic acid, which is either excreted, converted to carbon dioxide, or sequestered by the folate pathway through enzyme-catalysed condensation with THF. | ||
THF acts as a carrier of one-carbon units, which are attached to THF via nitrogen atoms at positions 5 and/or 10 (Fig. 1).1 The resultant THF derivatives are co-substrates for enzymes that catalyse the transfer of the one-carbon units to prime substrates. For example, the THF derivative 5,10-methylene-tetrahydrofolic acid (CH2-THF) provides the one-carbon unit for the methylation of deoxyuridine monophosphate (dUMP), which is catalysed by thymidylate synthase (TS, Fig. 1).8 This reaction, which forms thymidine monophosphate (dTMP), is essential for DNA replication and repair, and is consequently a target for cancer therapy.9
The major source of cellular CH2-THF is proposed to be serine metabolism, where serine hydroxymethyltransferases catalyse the transfer of the serinyl side-chain to THF (with concomitant conversion of serine to glycine).10 However, CH2-THF is also formed via the non-enzymatic condensation of THF with formaldehyde (HCHO), which is reportedly present in humans in micromolar concentrations (Fig. 1).11,12 Given the exposure of cells to HCHO from exogenous sources and via intracellular enzymatic demethylation reactions,13–15 it seems feasible that the reaction of THF and HCHO contributes to cellular CH2-THF levels. However, evidence for this is lacking. It is also unclear how quasi-stable CH2-THF can exist in the cellular environment where glutathione, a highly prevalent cellular thiol, reacts with HCHO and removes it from the cellular pool (Fig. 1).16,17 Recent pioneering work has additionally proposed that THF and derivatives are susceptible to degradation, interestingly releasing HCHO as a by-product.18 Therefore, THF degradation may provide HCHO for the folate pathway.
We are interested in defining the biochemistry of HCHO and are therefore interested in profiling its regulation of the folate pathway. To this end, we report NMR analyses on the complex biologically relevant chemistry of CH2-THF. Our results confirm HCHO-releasing degradation of THF but reveal CH2-THF to be stable to degradation under physiologically relevant conditions. This importantly suggests the presence of a novel non-enzymatic regulatory mechanism affecting folate stability and HCHO levels in cells.
Initially, we focused on confirming the production of HCHO during THF degradation, building on reported mass spectrometry studies.18 To identify HCHO production, a sample of THF stock (stored frozen to minimise prior degradation, Fig. S1 and S2, ESI†) was incubated in 50 mM ammonium formate buffer pH 7.5 (final concentration = 500 μM) for 20 hours at room temperature and then analysed by 1H NMR. Comparison of 1H NMR spectra before and after incubation revealed degradation of THF (Fig. S3 and S4, ESI†). However, the characteristic resonance for hydrated HCHO was not observed. However, it was noted that HCHO's dynamic reactivity with nucleophiles in the sample might result in signal broadening and loss of intensity; the hydrated HCHO resonance might also be obscured by overlap with the residual water resonance at δH 4.7 ppm.19 HCHO production was therefore assessed using the scavenging reagent 5,5-dimethylcyclohexane-1,3-dione (dimedone), which reacts with HCHO to form quasi-stable adducts detectable by their characteristic 1H NMR resonances.14 Repeating the experiment in the presence of dimedone (10 equivalents) revealed formation of two dimedone-HCHO adducts by 1H NMR analyses (Fig. S5, ESI†), suggesting HCHO is produced in the sample.
We then attempted to identify other degradation products. The formation of N-(4-aminobenzoyl)-L-glutamic acid (ABG) was evidenced by two characteristic doublet 1H NMR resonances at δH 6.87 ppm and δH 7.68 ppm (Fig. S3, ESI†), which were assigned to the aryl 4-aminobenzoyl protons on the basis of 1H–13C-HSQC analyses (which also suggested formation of HCHO, Fig. S4, ESI†) and by comparison with authentic ABG. While other 1H NMR resonances were observed in the spectra, their low intensities precluded assignment. However, analysis of authentic samples of folic acid and dihydrofolic acid (DHF) suggests these species were not formed (Fig. S6, ESI†). Importantly, only low-level resonances were observed in the aromatic region of the 1H NMR spectrum (except for ABG resonances), suggesting that any pterin-derived degradation products were either unstable, in dynamic exchange, or aggregating/precipitating from the sample. However, formation of pterin was evidenced by LC/MS analysis (calculated [M + H]+ = 164.0572 Da; detected 164.0575 Da). Therefore, it appears that THF degradation forms ABG, HCHO and pterin, supporting previous studies (Fig. 2A);18 however, other low-level products are possible.
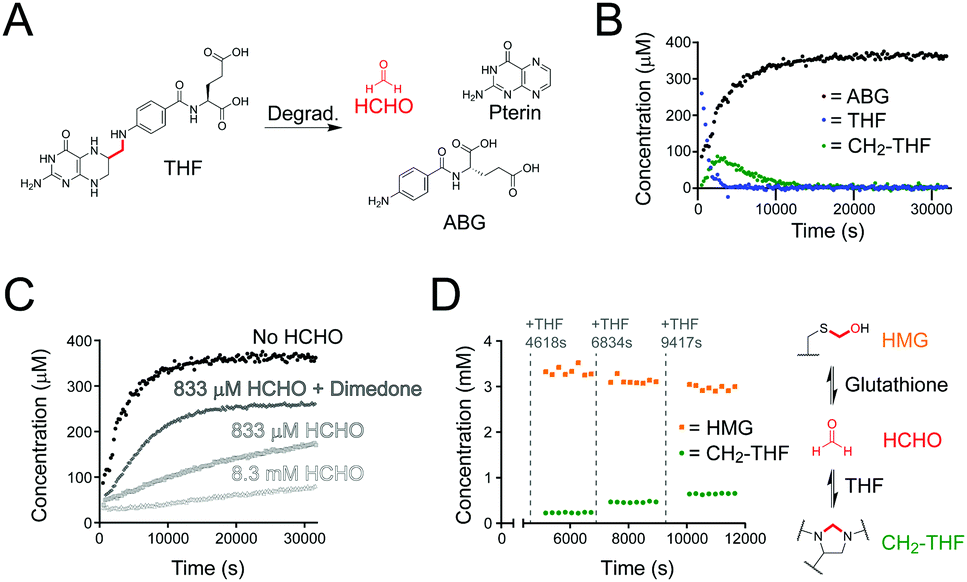 | ||
| Fig. 2 NMR studies on THF stability and reactivity with HCHO. (A) THF undergoes fragmentation under aqueous conditions to form HCHO (red), ABG and pterin. (B) NMR time-course of a sample of THF in ammonium formate buffer pH 7.5. The THF concentration decreased over time (blue), while the concentration of its degradation product ABG increased (black). In situ formation of CH2-THF (green) was observed at early time-points but its concentration decreased after 43 minutes. (C) ABG formation rates from samples of THF incubated with different concentrations of HCHO. The rate of ABG formation is slower when the free HCHO concentration is high. (D) NMR time-course monitoring the stability of HMG after exposure to THF. Initially, HCHO was reacted with an excess of glutathione to form HMG; THF was then added step-wise and aliquots of the reaction mixture were monitored by 1H NMR. Full conversion of THF to CH2-THF was observed (green), while the HMG concentration reduced (orange). | ||
We then determined the stability of the THF derivatives folic acid and CH2-THF under the same conditions. 1H NMR analyses of the samples after 16 hours incubation at room temperature revealed no degradation of folic acid; however, new products were present in the samples containing CH2-THF (analysed after 20 hours, Fig. S3, ESI†). The major identifiable product after CH2-THF degradation was the THF degradation product ABG; however, as CH2-THF in the sample was formed by the in situ reversible reaction of THF and HCHO, it was unclear whether ABG was formed via direct CH2-THF degradation, or from unreacted THF (for NMR characterisation of CH2-THF prepared with 13C-labelled HCHO, see Fig. S7 and S8, ESI†). Therefore, a time-course experiment was conducted on a sample containing THF and a small excess of HCHO (833 μM, 1.67 equivalents) to determine the kinetics of CH2-THF formation and stability. Interestingly, 1H NMR analysis revealed near-complete conversion of THF to CH2-THF from the first NMR time-point, suggesting THF reacts very quickly with HCHO under the tested conditions (Fig. S9A, ESI†). However, CH2-THF levels were observed to decrease after 18 minutes, while ABG levels increased at a reciprocal rate. Only trace levels of THF were observed during the analysis.
The formation and stability of CH2-THF was then determined in samples containing either no added HCHO, 8.3 mM HCHO (16.7 equivalents), or 833 μM HCHO with 5 mM dimedone (added last). As expected, the initial rate of CH2-THF formation was linearly dependent on the concentration of HCHO; however, CH2-THF was still observed in the sample without HCHO (Fig. 2B and Fig. S9, ESI†). In this sample, the CH2-THF level was low at early time-points but reached a maximum concentration of 87 μM after 43 minutes (Fig. 2B). After prolonged incubation, the CH2-THF concentration decreased, which coincided with formation of ABG. We then analysed the ABG formation rate in each sample. Interestingly, the ABG formation was inversely proportional to the HCHO concentration (Fig. 2C) – with no added HCHO, ABG formation was fastest (initial rate = 0.04 μM s−1), while at high HCHO concentration (8.3 mM), the ABG formation rate was markedly slower (0.0016 μM s−1) and remained constant throughout the analysis. ABG formation was also increased in the presence of excess dimedone (Fig. 2C). Therefore, these findings importantly imply that degradation to form ABG is slowed when THF reacts with HCHO. Given the reversibility of CH2-THF formation and its dependence on HCHO, the work suggests CH2-THF is either stable to degradation, or is at least markedly more stable than free THF.
We then monitored the formation and stability of CH2-THF in the presence of glutathione. Glutathione, which is present in high concentrations in cells, reacts reversibly with HCHO to form S-hydroxymethylglutathione (HMG), which is the substrate for the HCHO-metabolising enzyme glutathione-dependent alcohol dehydrogenase (ADH5, Fig. 1).16 Given the high cellular glutathione concentration and the efficiency of HMG formation,17 we were keen to determine whether quasi-stable CH2-THF can persist in a concentrated glutathione environment. We therefore conducted a time-course experiment with excess glutathione relative to THF (Fig. 2D). A stock solution of glutathione (20 mM) was first incubated with a relatively low concentration of HCHO (3.3 mM) in ammonium formate buffer to induce in situ formation of HMG (total volume = 3000 μL). An aliquot was then removed (582 μL) and supplemented with H2O (9 μL) and DMSO-d6 (9 μL), before analysis by 1H NMR. HMG formation was complete at the first time-point and remained constant throughout the analysis (65 min). While the free HCHO concentration after reaction could not be determined due to signal overlap, the excess of glutathione in the sample implies the free HCHO concentration is very low. Another aliquot was then removed from the stock sample (582 μL) and was supplemented with THF (6 μL of 50 mM THF in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 D2O:DMSO-d6), D2O (6 μL) and DMSO-d6 (6 μL). Time-course analysis of this aliquot revealed full conversion of the added THF to CH2-THF from the first time-point, while the HMG concentration decreased (Fig. 2D). The addition of THF to aliquots of the stock solution was then repeated twice more, with larger quantities of THF being added (12 μL and 18 μL of 50 mM THF in 1:1 D2O:DMSO-d6). This step-wise addition of THF also perturbed the equilibria, reducing the HMG concentration and resulting in full conversion of the added THF to CH2-THF (Fig. 2D). Therefore, this experiment reveals THF to be a significantly more efficient scavenger of HCHO than glutathione, and that HMG and CH2-THF are in dynamic exchange. The results also importantly suggest that CH2-THF can persist in concentrated glutathione environments, e.g. in cells.
:1 D2O:DMSO-d6), D2O (6 μL) and DMSO-d6 (6 μL). Time-course analysis of this aliquot revealed full conversion of the added THF to CH2-THF from the first time-point, while the HMG concentration decreased (Fig. 2D). The addition of THF to aliquots of the stock solution was then repeated twice more, with larger quantities of THF being added (12 μL and 18 μL of 50 mM THF in 1:1 D2O:DMSO-d6). This step-wise addition of THF also perturbed the equilibria, reducing the HMG concentration and resulting in full conversion of the added THF to CH2-THF (Fig. 2D). Therefore, this experiment reveals THF to be a significantly more efficient scavenger of HCHO than glutathione, and that HMG and CH2-THF are in dynamic exchange. The results also importantly suggest that CH2-THF can persist in concentrated glutathione environments, e.g. in cells.
We then studied CH2-THF formation in human cell lysate. HEK293F cells suspended in 50 mM ammonium formate buffer pH 7.5 (0.2 g mL−1) were lysed by sonication and centrifuged. A portion of the soluble fraction was then removed, mixed with D2O, and analysed by 1H NMR at 295 K. While multiple 1H resonances were observed in the 1H NMR spectrum, no evidence for THF or other folate derivatives was accrued, which suggests endogenous folate concentrations in these cells are too low for detection by 1H NMR (Fig. S10, ESI†). However, addition of THF to the lysate (1.5 mM final concentration) resulted in identifiable 1H resonances corresponding to THF (at δH 6.75 ppm and δH 7.55 ppm respectively, Fig. 3A). A third sample containing lysate, THF and an excess of 13C-labelled HCHO (13C-HCHO, 20-fold relative to added THF) was then prepared and analysed over 12 hours at 295 K. Interestingly, the THF-derived 1H resonances were observed to reduce in intensity from the first time-point (9 minutes), while new broad 1H resonances corresponding to CH2-THF emerged and persisted throughout the experiment (at δH 6.55 ppm and δH 7.65 ppm respectively, Fig. 3A). Formation of 13C-labelled CH2-THF was further implied by 1H–13C-HSQC analyses, which revealed HSQC correlations consistent with those expected for the 13C-labelled methylene (Fig. S11, ESI†). Overall, while these experiments were unable to confirm CH2-THF formation at endogenous concentrations, they imply that CH2-THF can be formed and can persist in the presence of cellular components. This is important because it suggests that the non-enzymatic reaction of HCHO and THF may contribute to cellular CH2-THF levels.
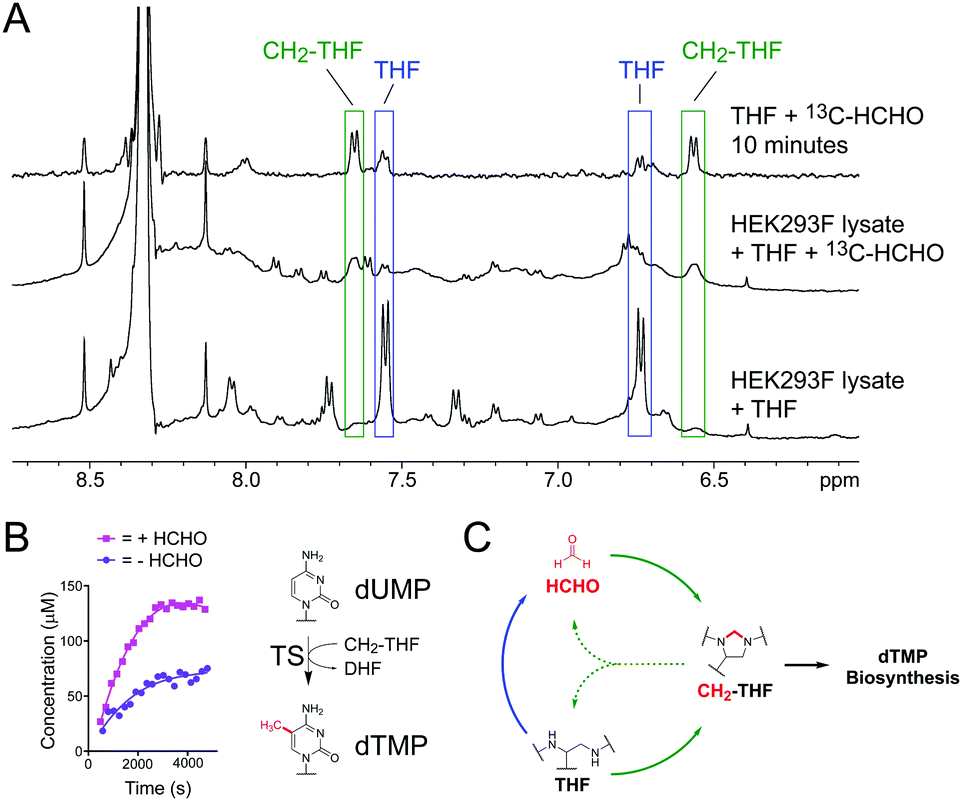 | ||
| Fig. 3 (A) 1H NMR spectra showing formation of CH2-THF in HEK293F cell lysate at 295 K. Increased intensities for characteristic CH2-THF 1H resonances were observed upon addition of 13C-HCHO (green). A 1H NMR spectrum in the absence of lysate is overlaid for comparison (conducted at 298 K). (B) NMR time-courses of TS-catalysed formation of dTMP in the presence (pink) or absence (purple) of added HCHO. Without added HCHO, some THF in the mixture degrades to form HCHO, which then reacts with remaining THF to form CH2-THF and promote catalysis. (C) HCHO mediates a feedback mechanism regulating the stability and reactivity of THF. THF reversibly reacts with HCHO to form CH2-THF (green arrows) but also degrades into HCHO when cellular HCHO levels are low (blue arrow). This HCHO can then react with residual THF to form the CH2-THF necessary for dTMP biosynthesis. | ||
We then conducted NMR activity assays on TS catalysis, which requires CH2-THF as a co-substrate (Fig. 1).8 Human recombinant His-tagged TS was prepared in Escherichia coli BL21 (DE3) cells and purified using a nickel affinity column (for TS expression and purification protocols, see ESI†). The enzyme was then incubated with substrate dUMP, THF and a small excess of HCHO (6.67 equivalents), before the mixture was transferred to an NMR tube and monitored by 1H NMR. Analysis of the 1H NMR spectra revealed formation of CH2-THF from the first time-point, suggesting that the added HCHO had reacted with THF. While enzymatic activity in the sample was low, the production of dTMP was evidenced by a new singlet resonance at δH 1.93 ppm, which was assigned to the methyl group of dTMP after addition of an authentic standard (Fig. S12, ESI†). There was also some evidence for the formation of the expected co-product DHF, although the low level of turnover precluded full assignment. The experiment was then repeated in the absence of HCHO; as expected, formation of dTMP was observed in the sample, albeit at a lower rate (Fig. 3B). Given that any CH2-THF in the sample is likely to have derived from THF degradation (either during the experiment or in the THF stock), the results imply that HCHO produced during THF degradation is sufficient to promote TS catalysis in vitro. It also can be rescued by THF degradation when HCHO levels are low.
Overall, our studies reveal that THF reacts efficiently with HCHO to form CH2-THF, but also degrades into HCHO under physiologically relevant aqueous conditions. Formation of CH2-THF is possible in the presence of glutathione and in human cell lysate, suggesting it is sufficiently stable to persist in cellular environments. Given the ubiquitous presence of HCHO in cells, it is therefore probable that the reaction between HCHO and THF contributes to cellular CH2-THF levels.
The observation that CH2-THF is more stable to degradation than THF is important because it implies the presence of a novel feedback mechanism (Fig. 3C). When cellular HCHO levels are high, THF is stabilised by forming CH2-THF, which both reduces HCHO production and sequesters HCHO from the cellular pool. This serves to protect cells from toxic HCHO overload. However, when HCHO levels are low, some unreacted THF degrades, thus providing the necessary HCHO for CH2-THF production and TS catalysis. While enzyme-mediated pathways will also regulate HCHO, THF and CH2-THF levels in cells, this non-enzymatic feedback mechanism suggests the presence of an additional cellular HCHO protection strategy mediated by THF. It also suggests that manipulation of cellular HCHO levels, e.g. by small molecules, can affect folate biology, thus implying new therapeutic strategies against metabolic and HCHO-mediated diseases. Finally, the observation that non-enzymatic chemistry between metabolites can affect their stability/biology has important implications for ongoing mechanistic studies in human metabolism, potentially across many pathways.
The work was funded by the Biochemical Society, UK. S. Y. C. acknowledges the Engineering and Physical Sciences Research Council for a studentship. The TS plasmid was a kind gift from Christopher Schofield (University of Oxford, UK), and the HEK293F cell pellet was a gift from Louise Fairall (University of Leicester, UK). We also thank Dr F. W. Muskett of the NMR facility within the Department of Molecular and Cell Biology/Leicester Institute of Structural and Chemical Biology for support with NMR time-course experiments. We also thank Vanessa Timmermann and Sharad Mistry for help with NMR and mass spectrometry characterisation studies respectively.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. S. Ducker and J. D. Rabinowitz, Cell Metab., 2017, 25, 27 CrossRef CAS PubMed.
- P. Goyette, P. Frosst, D. S. Rosenblatt and R. Rozen, Am. J. Hum. Genet., 1995, 56, 1052 CAS.
- J. P. Casas, L. E. Bautista, L. Smeeth, P. Sharma and A. D. Hingorani, Lancet, 2005, 365, 224 CrossRef CAS.
- A. E. Beaudin and P. J. Stover, Birth Defects Res., Part A, 2009, 85, 274 CrossRef CAS PubMed.
- A. J. A. Wright, J. R. Dainty and P. M. Finglas, Br. J. Nutr., 2007, 98, 667 CrossRef CAS PubMed.
- D. Fernández-Villa, M. R. Aguilar and L. Rojo, Int. J. Mol. Sci., 2019, 20, 4996 CrossRef PubMed.
- N. Hagner and M. Joerger, Cancer Manage. Res., 2010, 2, 293 CAS.
- C. W. Carreras and D. V. Santi, Annu. Rev. Biochem., 1995, 64, 721 CrossRef CAS PubMed.
- K. Takezawa, I. Okamoto, S. Tsukioka, J. Uchida, M. Kiniwa, M. Fukuoka and K. Nakagawa, Br. J. Cancer, 2010, 103, 354 CrossRef CAS PubMed.
- A. Tramonti, C. Nardella, M. L. di Salvo, A. Barile, F. Cutruzzolà and R. Contestabile, Biochemistry, 2018, 57, 6984 CrossRef CAS PubMed.
- H. He, E. Noor, P. A. Ramos-Parra, L. E. García-Valencia, J. A. Patterson, R. I. Díaz de la Garza, A. D. Hanson and A. Bar-Even, Metabolites, 2020, 10, 65 CrossRef CAS PubMed.
- European Food Safety Authority, EFSA J., 2014, 12, 3550 Search PubMed.
- J. Wei, F. Liu, Z. Lu, Q. Fei, Y. Ai, P. C. He, H. Shi, X. Cui, R. Su, A. Klungland, G. Jia, J. Chen and C. He, Mol. Cell, 2018, 71, 973 CrossRef CAS PubMed.
- D. H. Porter, R. J. Cook and C. Wagner, Arch. Biochem. Biophys., 1985, 243, 396 CrossRef CAS PubMed.
- L. J. Walport, R. J. Hopkinson and C. J. Schofield, Curr. Opin. Chem. Biol., 2012, 16, 525 CrossRef CAS PubMed.
- N. Harms, J. Ras, W. N. M. Reijnders and R. J. M. van Spanning, J. Bacteriol., 1996, 178, 6296 CrossRef CAS PubMed.
- R. J. Hopkinson, P. S. Barlow, C. J. Schofield and T. D. W. Claridge, Org. Biomol. Chem., 2010, 8, 4915 RSC.
- G. Burgos-Barragan, N. Wit, J. Meiser, F. A. Dingler, M. Pietzke, L. Mulderrig, L. B. Pontel, I. V. Rosado, T. F. Brewer, R. L. Cordell, P. S. Monks, C. J. Chang, A. Vazquez and K. J. Patel, Nature, 2017, 548, 612 CrossRef CAS PubMed.
- M. Rivlin, U. Eliav and G. Navon, J. Phys. Chem. B, 2015, 119, 4479 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for production of recombinant thymidylate synthase and NMR analyses. See DOI: 10.1039/d1cc01425k |
| This journal is © The Royal Society of Chemistry 2021 |