
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA monomeric methyllithium complex: synthesis and structure†‡
Nathan
Davison§
,
Emanuele
Falbo§
 ,
Paul G.
Waddell
,
Thomas J.
Penfold
and
Erli
Lu
*
,
Paul G.
Waddell
,
Thomas J.
Penfold
and
Erli
Lu
*
Chemistry-School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: erli.lu@newcastle.ac.uk
First published on 20th May 2021
Abstract
Methyllithium (MeLi) is the parent archetypal organolithium complex. MeLi exists as aggregates in solutions and solid states. Monomeric MeLi is postulated as a highly reactive intermediate and plays a vital role in understanding MeLi-mediated reactions but has not been isolated. Herein, we report the synthesis and structure of the first monomeric MeLi complex enabled by a new hexadentate neutral amine ligand.
Since the pioneering work by Wilhelm Schlenk and Joanna Holtz in 1917,1 organolithium complexes (RLi; R: alkyl, aryl, alkenyl, and alkynyl) have enabled numerous organic, inorganic, organometallic and polymerization reactions, acting as the cornerstone of organometallic chemistry,2–5 and are still an active research frontier.6 RLi complexes exist as aggregates in solutions and solid states.7–11 On the one hand, these aggregates stabilise the highly polar and reactive Li–C bonds; but in general, these aggregates also deactivate the RLi reagents.12 Breaking the aggregates into the corresponding RLi monomer is postulated as the key step in RLi-mediated reactions.7 Therefore, the synthesis and structural studies of RLi monomers are of crucial importance to understand the mechanism of these RLi-mediated reactions and are hence germane to physical organic and organometallic communities.
To break RLi aggregates, Lewis basic ligands, such as amines and ethers, are necessary.8 Ligand-supported RLi monomers have been pursued by coordination chemists for decades, but there is still a significant knowledge gap. All such reported isolable RLi monomers are stabilised by sterically bulky alkyls (R), i.e. –CH2SiMe3,12 –CH(SiMe3)2,13,16tert-butyl,14,15 sec-butyl16 and isopropyl,17 electronically delocalised aryls18–21/benzyls,22 or heteroatom-substituted alkyls.23 Methyllithium (MeLi) is the parent archetypal member of the RLi family,1,24,25 which has been widely used as a methyl synthon, a nucleophilic reagent and a strong Brønsted base.26 However, to the best of our knowledge, an isolable ligand-supported MeLi monomer is unknown.
Though a ligand-free MeLi monomer has been observed in an inert gas matrix27 or as a short-lived species in sub-millimeter spectroscopy,28 all previous attempts to synthesize ligand-supported MeLi monomers resulted in dimers. For instance, the N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA) and (R,R)-N,N,N′,N′-tetramethyl-1,2-diaminocyclohexane [(R,R)-TMCDA] supported MeLi dimers were reported by Strohmann and co-workers in 200716 and 2020,29 respectively (Chart 1a and b). The state-of-the-art bi- and tri-dentate ligands, such as [(R,R)-TMCDA] and PMDTA, which though working for bulkier alkyls (e.g. tert-butyl), are proven to be insufficient to stabilize the MeLi monomer against dimerization. In this work, with a bespoke hexadentate ligand, N,N′,N′′-tris-(2-N-diethylaminoEthyl)-1,4,7-triAza-cycloNonane (DETAN) (1), we report the first isolable monomeric MeLi complex [Li(CH3)(κ3-N,N′,N′′-DETAN)] (2) (Chart 1c).
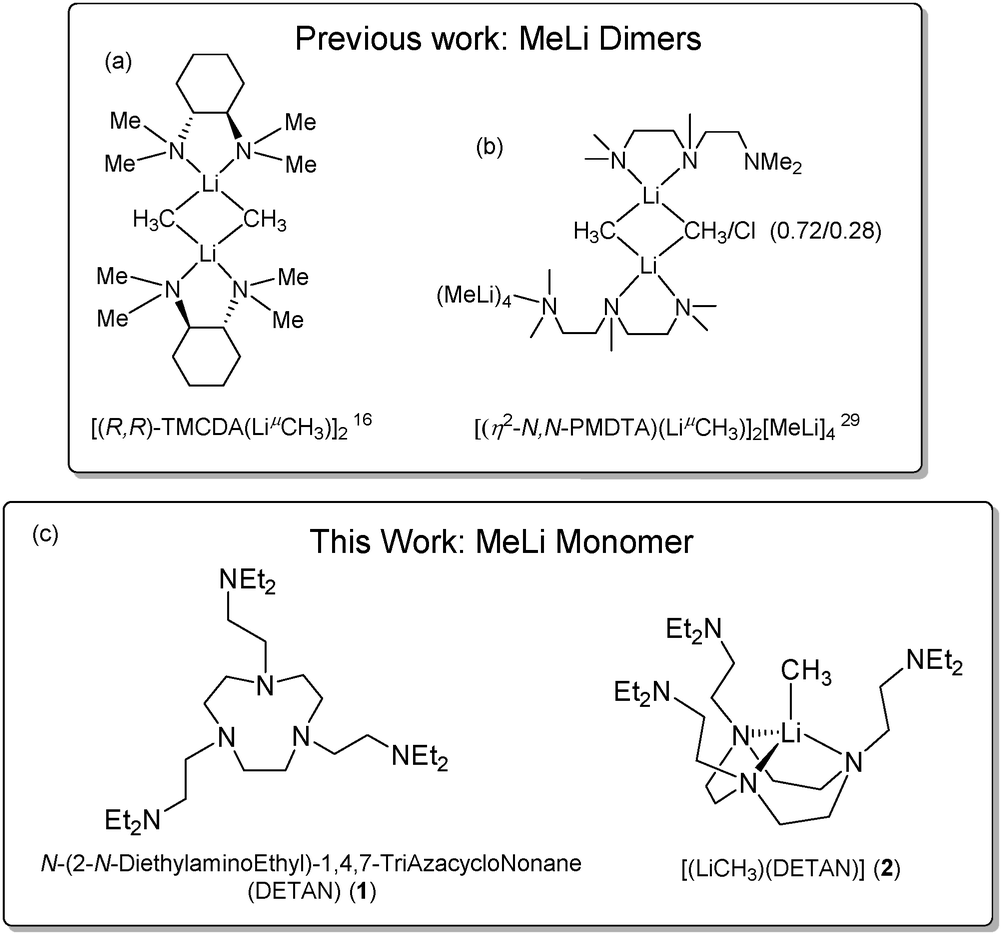 | ||
| Chart 1 (a and b) Previous studies on methyllithium dimers.16,29 (c) This work: a hexadentate ligand DETAN (1) and the DETAN-supported methyllithium monomer (2). | ||
The DETAN ligand 1 is designed and synthesized (Scheme 1) by combining a rigid macrocyclic backbone and three flexible pendant arms. Meanwhile, 1 is composed of only inert C–H, C–C and N–C binds to avoid ligand lithiation.30,31 These features are essential to stabilize highly reactive species such as the MeLi monomer. The following reaction between 1 and MeLi requires a careful design. For example, cyclic ethers such as tetrahydrofuran (THF) must be avoided due to their liability of C–H activation and ring-opening decomposition with alkyllithium reagents.32,33 Gratifyingly, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction between 1 and MeLi (1.6 M in Et2O) in hexane/Et2O (20/1) mixed solvents at lower temperatures (−80 to −30 °C) produced the desired MeLi monomer [Li(CH3)(κ3-N,N′,N′′-DETAN)] (2) as colourless crystals in 25% yield (Scheme 1). Complex 2 is stable as colourless crystals under −20 °C for several days but decomposes at room temperature within 15 minutes, possibly via the α- or β-deprotonation of the DETAN ligand (similar to the PMDTA/TMCDA ligands17,31). Once crystallized, complex 2 is insoluble in aliphatic solvents and decomposes in ethereal and aromatic solvents, e.g. THF and toluene, at −20 °C. The instability prevents us from obtaining reliable NMR spectra and CHN elemental analysis data. It is noteworthy that similar experimental challenges were reported in the PMDTA-supported MeLi dimer,29 [(R,R)-TMCDA]-supported iPrLi dimer17 and sBuLi monomer.17
:1 reaction between 1 and MeLi (1.6 M in Et2O) in hexane/Et2O (20/1) mixed solvents at lower temperatures (−80 to −30 °C) produced the desired MeLi monomer [Li(CH3)(κ3-N,N′,N′′-DETAN)] (2) as colourless crystals in 25% yield (Scheme 1). Complex 2 is stable as colourless crystals under −20 °C for several days but decomposes at room temperature within 15 minutes, possibly via the α- or β-deprotonation of the DETAN ligand (similar to the PMDTA/TMCDA ligands17,31). Once crystallized, complex 2 is insoluble in aliphatic solvents and decomposes in ethereal and aromatic solvents, e.g. THF and toluene, at −20 °C. The instability prevents us from obtaining reliable NMR spectra and CHN elemental analysis data. It is noteworthy that similar experimental challenges were reported in the PMDTA-supported MeLi dimer,29 [(R,R)-TMCDA]-supported iPrLi dimer17 and sBuLi monomer.17
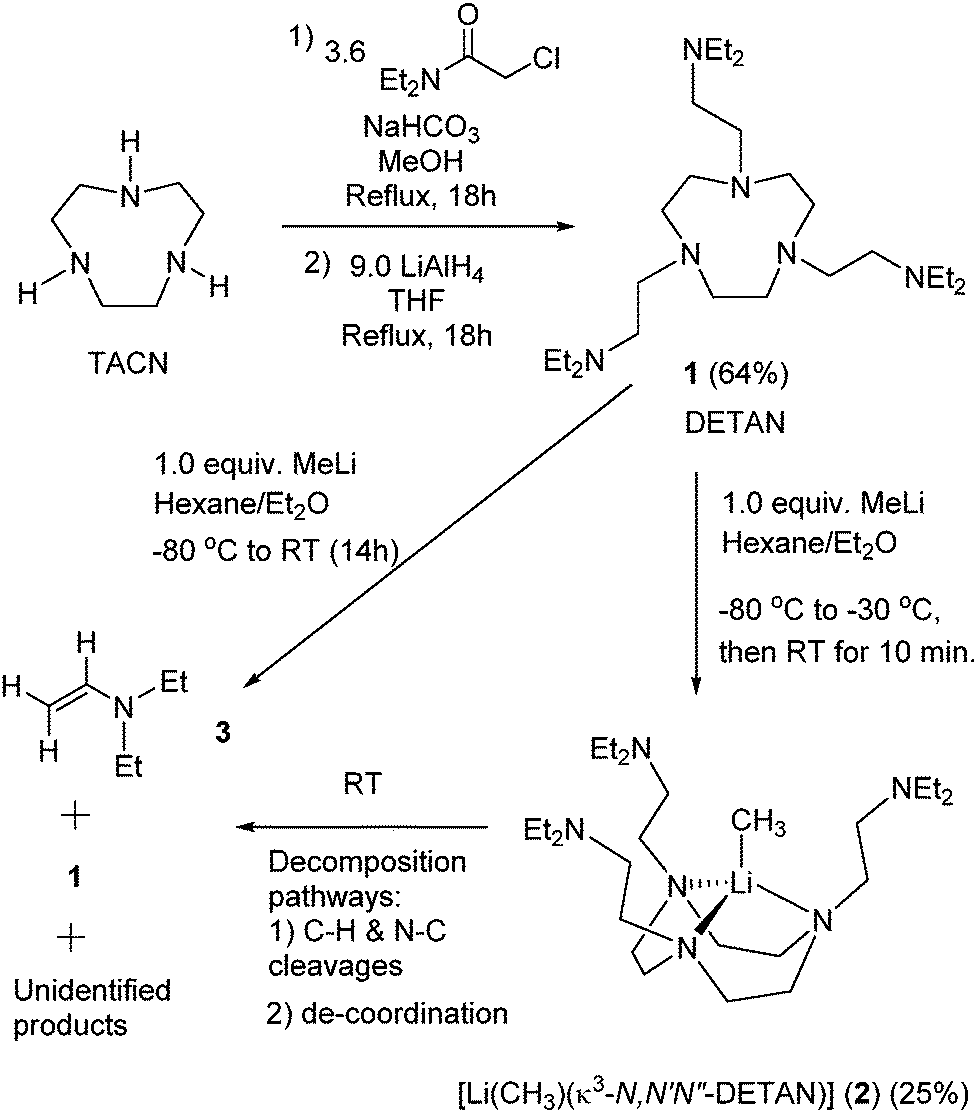 | ||
| Scheme 1 Synthesis of the N,N-diethyl DETAN ligand (1) and the methyllithium monomer [Li(CH3)(κ3-N,N′,N′′-DETAN)] (2). | ||
The instability of 2 was rationalised by a room temperature reaction between 1 and MeLi (Scheme 1). Instead of 2 from the low-temperature reaction, a complex mixture was obtained, among which N,N-diethyl ethenamine (3)34,35 and 1 can be identified using the 1H NMR spectrum (Fig. S3, ESI‡). It should be noted that the mixture is heterogeneous, containing a minor amount of white solid insoluble in d6-benzene, which is possibly a mixture of [MeLi]n and other decomposition products. At least two decomposing pathways can be postulated: (1) C–H and N–C cleavages to produce 3 and (2) de-coordination to produce [MeLi]n and 1 (Fig. S3, ESI‡). Similar ligand C–H and N–C cleavages were reported in a tBuLi monomer30 and a scandium imido complex36 but not in the MeLi dimers.16,29 In general, MeLi was considered to be less reactive than tBuLi and nBuLi.7–10 However, the facile decomposition of 2 suggests that the MeLi reactivity is enhanced by forming a monomer.
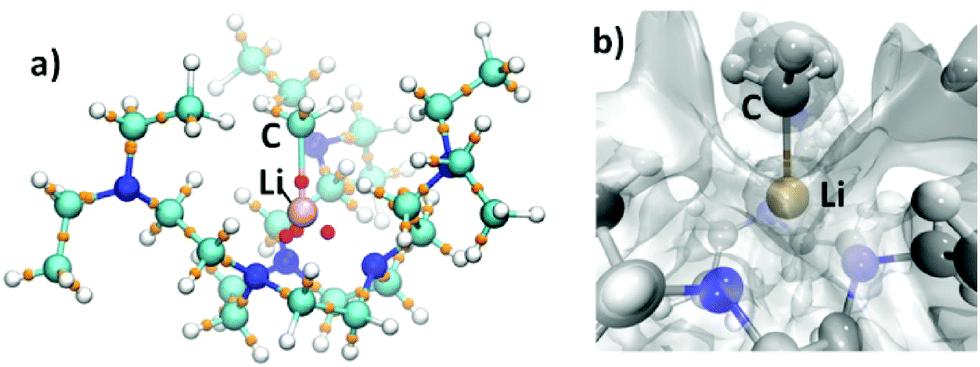
The solid state structure of complex 2 is elucidated by single-crystal X-ray diffraction (SCXRD), which confirms its monomeric nature (Fig. 1). Complex 2 crystallises in the trigonal P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group with the Li1–C1 bond orientated along a three-fold axis such that only one-third of the molecule is crystallographically independent. The most salient structural feature of 2 is the monomeric MeLi unit. Unlike the higher MeLi aggregates,25 there are no intermolecular Me⋯Li interactions in 2. In fact, the intermolecular interactions in the solid state packing structure of 2 are dominated by van der Waals and dispersion forces without observable hydrogen bonds or Me⋯Li interactions. The methyl group is terminally coordinated to the Li centre, which is at the short end of all reported Li–C bond lengths. The Li1–C1 bond in 2 is at the short end of all reported Li–C bond lengths.9,37 It is much shorter than the Li–μCH3 bonds in the previous MeLi dimers (2.18–2.28 Å),16,29 reflecting the monomeric nature of 2, and is slightly shorter than the Li–CtBu bond (2.114(4) Å) in a [Li(tBu)(−)-sparteine] monomer.14 The Li1–C1 bond length in 2 is only superseded by the [Li(tBu) (R,R-TMCDA)] monomer (Li–CtBu = 2.064(15) Å), which is likely a result of the relatively small (R,R-TMCDA) ligand.16 Besides the methyl group, the Li centre is coordinated by three neutral nitrogen atoms (N1A) in the macrocyclic ring via the Li ← NRing dative bonds (2.106(4) Å), forming a Li-centred tetrahedral geometry. The macrocyclic backbone provides the essential structural rigidity to support the monomeric MeLi unit. Meanwhile, the pendant arms are coordination-free but play a vital role in stabilizing the monomeric MeLi unit by forming a protected cavity. This is clearly illustrated by the space-filling model (top-view and side-view along the Li1–C1 bond) of complex 2 (Fig. 1b and c). In comparison, the N,N′,N′′-trimethyl-1,3,5-triazacyclononane (Me3TACN) ligand, which bears a similar macrocyclic TACN backbone but does not have the pendant arms, was reported to form a tert-butyllithium trimer [(tBuLi)3(Me3TACN)].38
space group with the Li1–C1 bond orientated along a three-fold axis such that only one-third of the molecule is crystallographically independent. The most salient structural feature of 2 is the monomeric MeLi unit. Unlike the higher MeLi aggregates,25 there are no intermolecular Me⋯Li interactions in 2. In fact, the intermolecular interactions in the solid state packing structure of 2 are dominated by van der Waals and dispersion forces without observable hydrogen bonds or Me⋯Li interactions. The methyl group is terminally coordinated to the Li centre, which is at the short end of all reported Li–C bond lengths. The Li1–C1 bond in 2 is at the short end of all reported Li–C bond lengths.9,37 It is much shorter than the Li–μCH3 bonds in the previous MeLi dimers (2.18–2.28 Å),16,29 reflecting the monomeric nature of 2, and is slightly shorter than the Li–CtBu bond (2.114(4) Å) in a [Li(tBu)(−)-sparteine] monomer.14 The Li1–C1 bond length in 2 is only superseded by the [Li(tBu) (R,R-TMCDA)] monomer (Li–CtBu = 2.064(15) Å), which is likely a result of the relatively small (R,R-TMCDA) ligand.16 Besides the methyl group, the Li centre is coordinated by three neutral nitrogen atoms (N1A) in the macrocyclic ring via the Li ← NRing dative bonds (2.106(4) Å), forming a Li-centred tetrahedral geometry. The macrocyclic backbone provides the essential structural rigidity to support the monomeric MeLi unit. Meanwhile, the pendant arms are coordination-free but play a vital role in stabilizing the monomeric MeLi unit by forming a protected cavity. This is clearly illustrated by the space-filling model (top-view and side-view along the Li1–C1 bond) of complex 2 (Fig. 1b and c). In comparison, the N,N′,N′′-trimethyl-1,3,5-triazacyclononane (Me3TACN) ligand, which bears a similar macrocyclic TACN backbone but does not have the pendant arms, was reported to form a tert-butyllithium trimer [(tBuLi)3(Me3TACN)].38
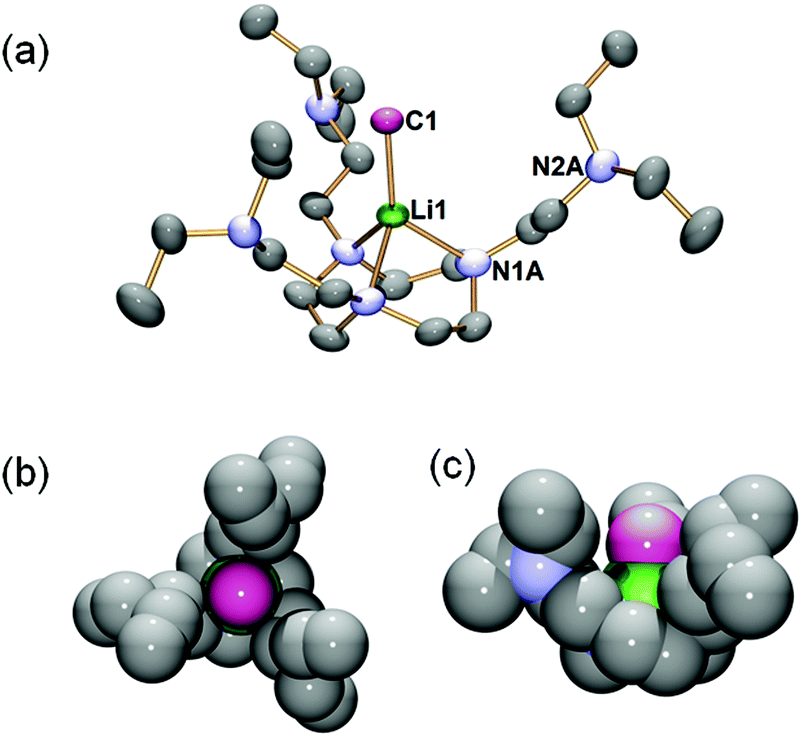 | ||
| Fig. 1 (a) X-ray crystal structure of [Li(CH3)(κ3-N,N′N′′-DETAN)] (2) at 150 K with 50% probability ellipsoids. Hydrogen atoms are omitted and only the orientation of the macrocycle with the highest occupancy is shown for clarity. Only atoms of the crystallographically independent fragment are labelled. (b and c) Space filling presentation for 2, top-view (b) and side-view (c) against the Li1–C1 bond, respectively (grey: carbon; green: lithium; blue: nitrogen). The selected bond distances (Å) and angles (°) are Li1–C1, 2.099(5); Li1–N1A, 2.106(4); C1–Li1–N1A, 127.62(12); N1A–Li1–N1A, 86.63(18). The atomic colour codes: Li (green); C (excluding Li–CH3) (grey); Li–CH3 (fuchsia); N (blue). | ||
Geometry optimisations using density functional theory (DFT) calculations within the approximation of the ωB97x-D339 exchange and correlation functional and a def2-TZVP40 basis set were conducted to gain structural insights into the MeLi monomer 2. The bond lengths and angles obtained compared well to those obtained using SCXRD (Fig. S5, ESI‡) (Table S3, ESI‡), e.g. Li–CH3 2.069 Å (calculated) cf. 2.099(5) Å (SCXRD). The nature of the Li–C bond in alkyllithium complexes, particularly in the parent MeLi, is a long running debate in the theoretical chemistry community; however, most of the previous studies were based on a hypothetic gas-phase monomeric MeLi molecule.41–49 Complex 2 provides an unprecedented opportunity to probe the Li–C bonding scenario in a synthesized monomeric MeLi molecule.
Natural population analysis (NPA) (ωB97x-D339/def2-TZVP40) gives a Li atomic charge of +0.451, while the CMe atomic charge is −1.313. The Li NPA atomic charge of +0.451 may suggest some Li–C bond covalency;42–44 however, atoms-in-molecule (AIM) analysis gives a Li and CMe atomic charges of +0.854 and −0.400, respectively (Table S4, ESI‡). Caution must be taken when interpreting the bonding character using atomic charge values alone: as shown, different methods can give rise to very different values.42 Thus, orbital and topology analyses are conducted to gain an overview of the Li–C bonding picture.
Natural bonding orbital (NBO) analysis (ωB97x-D336/def2-TVZPP37) returns a highest occupied molecular orbital (HOMO) as a σ bonding orbital (Fig. 2) localised between Li and CH3, with a major carbon character (66%, among which 4.2% s and 61.8% p) but also a non-negligible lithium contribution (8.8%, among which 2.7% s and 6.1% p). The donor–acceptor interaction analysis of NBO was carried out by examining all possible interactions between ‘filled’ Lewis-type NBOs (donors) and ‘empty’ non-Lewis NBOs (acceptors) and estimating their energetic significance using the 2nd-order perturbation theory. It was found that the lone pair anti-bonding (LP*) of the Li atom and the lone pair (LP) of CMe give the strongest stabilisation energy in 2 (78.87 kcal mol−1), compared to much weaker N → Li dative interactions (11.97 kcal mol−1). These results clearly suggest that the Li–CMe bond in 2 is not purely ionic (electrostatic) in nature but with non-negligible orbital overlapping and substantial electron density sharing.
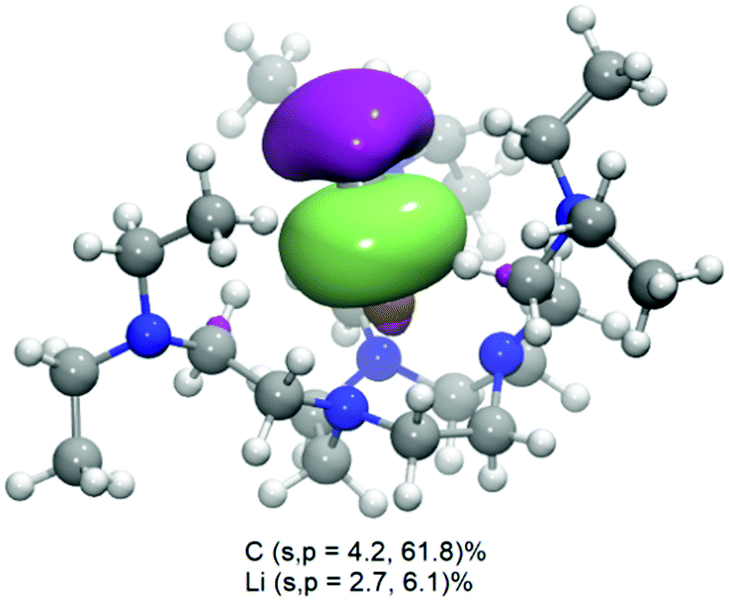 | ||
| Fig. 2 The Kohn–Sham highest occupied molecular orbital (HOMO) of complex 2 at the ωB97x-D336/def2-TVZP37 level of theory. MOs isovalue = 0.03. The percentage of atomic orbital (AOs) contribution to the HOMO was computed through the NBO analysis. | ||
Despite the NBO analysis suggesting evidence for Li–C covalency, to address the discrepancy between the NPA and AIM atomic charge calculations, we conducted quantum theory of atoms in molecules (QTAIM) and electron localisation function (ELF) analyses. Both of these analyses were conducted with the electron density obtained from DFT (ωB97x-D339). The combination of QTAIM and ELF methods has been successfully used to address the bonding covalency/ionicity problem.50,51 For 2, the QTAIM52 analysis returned a Li–C bond critical point (BCP) closer to the Li atom than to the CMe atom (Fig. 3a), with the electron density (ρ) and Laplacian (∇2ρ) values (a.u.) at BCPs of 0.034 and 0.134, respectively. The values of 0.036 (ρ) and 0.202 (∇2ρ) for the Li–CH3 bonds were obtained from coupled cluster single-double (CCSD) calculations. These values, in agreement with the AIM atomic charge values, suggest a highly ionic Li–C bond. To corroborate the relevance between the QTAIM calculation and the Li–C bonding scenario,53 we conducted ELF analysis, which suggests that the electron density largely remains on the CMe atom (Fig. 3b).
 | ||
| Fig. 3 (a) Graphical representation of (3,−1) BCPs (red) involving the Li atom and all other remaining BCPs (orange) from the QTAIM analysis of complex 2 carried out on the CCSD electron density. All other nucleus (3,−3), attractor (3,−3), ring (3+1), and cage (3+3) CPs are not shown for clarity. (b) The ELFs of complex 2, focusing on the Li–CH3 unit. Isovalue = 0.65. | ||
In conclusion, using a hexadentate neutral ligand DETAN (1), we were able to isolate the parent organolithium, methyllithium, in its monomeric form. The DETAN-supported MeLi monomer, [Li(CH3)(κ3-N,N′N′′-DETAN)] (2), features a short terminal Li–C bond (2.099(5) Å), which was found to be predominantly ionic and polarised but with a non-negligible orbital overlap-driven covalency. Further reactivity studies of 2, as well as those on its heavier group 1 metal congeners, are underway.
N. D. and E. F. contributed equally by conducting synthesis and characterisation (N. D.) and calculations (E. F., under T. J. P.’s supervision), respectively. P. G. W. collected, solved, refined and analysed the crystallographic data. E. L. conceptualised the central idea, supervised the work, analysed the data and wrote the manuscript with contributions from all the authors.
N. D. and E. L. thank Professor Andrew C. Benniston and Dr Keith Izod (both are from Newcastle University) for their support in laboratory infrastructure. The authors thank Professor Yaofeng Chen (Shanghai Institute of Organic Chemistry-Chinese Academy of Sciences) and Professor Stephen T. Liddle (University of Manchester) for their insightful discussions. E. L. acknowledges the Newcastle University Academic Track (NUAcT) Fellowship and The Royal Society of Chemistry Research Enablement Grants (E20-5153) for financial support. T. J. P. thanks the EPSRC for funding (EP/R021503/1). The authors thank the Faculty of Science, Agriculture and Engineering (SAgE) Mass Spectrometry Facility at Newcastle University for the mass spectroscopy service.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- W. Schlenk and J. Holtz, Ber. Dtsch. Chem. Ges., 1917, 50, 262 CrossRef CAS.
- U. Wietelmann and J. Klett, Z. Anorg. Allg. Chem., 2018, 644, 194 CrossRef CAS PubMed.
- In The chemistry of organolithium compounds, ed. Z. Rappoport and I. Mare, John Wiley & Sons, Ltd, Chichester, West Sussex, England, 2004 Search PubMed.
- V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204 RSC.
- T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247 CrossRef CAS PubMed.
- J. J. Gladfelder, S. Ghosh, M. Podunavac, A. W. Cook, Y. Ma, R. A. Woltornist, I. Keresztes, T. W. Hayton, D. B. Collum and A. Zakarian, J. Am. Chem. Soc., 2019, 141, 15024 CrossRef CAS PubMed.
- V. H. Gessner, C. Däschlein and C. Strohmann, Chem. – Eur. J., 2009, 15, 3320 CrossRef CAS PubMed.
- H. J. Reich, Chem. Rev., 2013, 113, 7130 CrossRef CAS PubMed.
- A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470 CrossRef CAS PubMed.
- F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563 CrossRef CAS PubMed.
- R. A. Gossage, J. T. B. H. Jatrzebski and G. van Koten, Angew. Chem., Int. Ed., 2005, 44, 1448 CrossRef CAS PubMed.
- But the trend is not without exception. See: L. Knauer, J. Wattenberg, U. Kroesen and C. Strohmann, Dalton Trans., 2019, 48, 11285 RSC.
- M. F. Lappert, L. M. Engelhardt, C. L. Raston and A. H. White, J. Chem. Soc., Chem. Commun., 1982, 1323 RSC.
- C. Strohmann, T. Seibel and K. Strohfeldt, Angew. Chem., Int. Ed., 2003, 42, 4531 CrossRef CAS PubMed.
- C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 8281 CrossRef CAS PubMed.
- C. Strohmann and V. H. Gessner, J. Am. Chem. Soc., 2007, 129, 8952 CrossRef CAS PubMed.
- C. Strohmann, V. H. Gessner and A. Damme, Chem. Commun., 2008, 3381 RSC.
- T. Maetzke and D. Seebach, Helv. Chim. Acta, 1989, 72, 624 CrossRef CAS.
- G. S. Girolami, M. E. Riehl, K. S. Suslick and S. R. Wilson, Organometallics, 1992, 11, 3907 CrossRef CAS.
- R. C. Crittendon, B. C. Beck, J. Su, X.-W. Li and G. H. Robinson, Organometallics, 1999, 18, 156 CrossRef CAS.
- N. J. Hardmann, B. Twamley, M. Stender, R. Baldwin, S. Hino, B. Schiemenz, S. M. Kauzlarich and P. P. Power, J. Organomet. Chem., 2002, 643–644, 461 CrossRef.
- M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 3364 CrossRef CAS PubMed.
- H. H. Karsch, K. Zellner, P. Mikulcik, J. Lachmann and G. Müller, Organometallics, 1990, 9, 190 CrossRef CAS.
- T. L. Brown and M. T. Rogers, J. Am. Chem. Soc., 1957, 79, 1859 CrossRef CAS.
- E. Weiss and E. A. C. Lucken, J. Organomet. Chem., 1964, 2, 197 CrossRef CAS.
- M. J. Lusch, W. V. Philips, R. F. Sieloff, G. S. Nomura and H. O. House, Org. Synth., 1984, 62, 101 CrossRef CAS.
- L. Andrews, J. Chem. Phys., 1967, 47, 4834 CrossRef CAS.
- D. B. Grotjahn, T. C. Pesch, J. Xin and L. M. Ziurys, J. Am. Chem. Soc., 1997, 119, 12368 CrossRef CAS.
- L. Knauer and C. Strohmann, Chem. Commun., 2020, 56, 13543 RSC.
- V. H. Gessner and C. Strohmann, J. Am. Chem. Soc., 2008, 130, 14412 CrossRef CAS PubMed.
- C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 4566 CrossRef CAS PubMed.
- A. R. Kennedy, J. Klett, R. E. Mulvey and D. S. Wright, Science, 2009, 326, 706 CrossRef CAS PubMed.
- R. E. Mulvey, V. L. Blair, W. Clegg, A. R. Kennedy, J. Klett and L. Russo, Nat. Chem., 2010, 2, 588 CrossRef CAS PubMed.
- P. L. F. Chang and D. C. Dittmer, J. Org. Chem., 1969, 34, 2791 CrossRef CAS.
- X. Zhang, A. Fried, S. Knapp and A. S. Goldman, Chem. Commun., 2003, 2060 RSC.
- J.-X. Chu, Q.-H. Zhou, Y.-X. Li, X.-B. Leng and Y.-F. Chen, Sci. China: Chem., 2014, 57, 1098 CrossRef CAS.
- A. Münch, L. Knauer, H. Ott, C. Sindlinger, R. Herbst-Irmer, C. Strohmann and D. Stalke, J. Am. Chem. Soc., 2020, 142, 15897 CrossRef PubMed.
- C. Strohmann and V. H. Gessner, Chem. – Asian J., 2008, 3, 1929 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- W. Florian, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- A. Stretwiesser, S. M. Bachrach, A. Dorigo and P. V. R. Schleyer, In Lithium Chemistry. A Theoretical and Experimental Overview, ed. A.-M. Sapse, P. V. R. Schleyer, J. Wiley & Sons, New York, 1995, Ch. 1 Search PubMed.
- F. M. Bickelhaupt, M. Solà and C. F. Guerra, J. Chem. Theory Comput., 2006, 2, 965 CrossRef CAS PubMed.
- F. M. Bickelhaupt, N. J. R. v. E. Hommes, C. F. Guerra and E. J. Baerends, Organometallics, 1996, 15, 2923 CrossRef CAS.
- F. M. Bickelhaupt, M. Solà and C. F. Guerra, J. Mol. Model., 2006, 12, 563 CrossRef CAS PubMed.
- A. Streitwieser, Jr., J. E. Williams, S. Alexandratos and J. M. McKelvey, J. Am. Chem. Soc., 1976, 98, 4778 CrossRef.
- G. Graham, S. Richtsmeier and D. A. Dixon, J. Am. Chem. Soc., 1980, 102, 5759 CrossRef CAS.
- E. Kaufmann, K. Raghavachari, A. E. Reed and P. V. R. Schleyer, Organometallics, 1988, 7, 1597 CrossRef CAS.
- K. B. Wiberg and C. M. Breneman, J. Am. Chem. Soc., 1990, 112, 8765 CrossRef CAS.
- R. Ponec, J. Roithová, X. Gironés, L. Lain, A. Torre and R. Bochicchio, J. Phys. Chem. A, 2002, 106, 1019 CrossRef CAS.
- M. Gregson, E. Lu, F. Tuna, E. J. L. McInnes, C. Hennig, A. C. Scheinost, J. McMaster, W. Lewis, A. J. Blake, A. Kerridge and S. T. Liddle, Chem. Sci., 2016, 7, 3286 RSC.
- E. Lu, S. Sajjad, V. E. J. Berryman, A. J. Wooles, N. Kaltsoyannis and S. T. Liddle, Nat. Commun., 2019, 10, 634 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed.
- T. Clark, J. S. Murray and P. Politzer, Phys. Chem. Chem. Phys., 2018, 20, 30076 RSC.
Footnotes |
| † This Communication is dedicated to the memory of Professor Hans J. Reich (University of Wisconsin) for his pioneering work in physical organometallic chemistry. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2064390. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc01420j |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |