
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and synthesis of gene-directed caged cyclic nucleotides exhibiting cell type selectivity†
Akinobu Z.
Suzuki‡
a,
Taichi
Sakano‡
a,
Hirona
Sasaki
a,
Rei
Watahiki
a,
Masaki
Sone
a,
Kazuki
Horikawa
b and
Toshiaki
Furuta
 *a
*a
aDepartment of Biomolecular Science, Faculty of Science, Toho University, 2-2-1 Miyama, Funabashi, 274-8510, Japan. E-mail: furuta@biomol.sci.toho-u.ac.jp; Fax: +81 47-472-1169
bDepartment of Optical Imaging, Institute of Biomedical Sciences, Tokushima University Graduate School, 3-18-15 Kuramoto Cho, Tokushima City, Tokushima 770-8503, Japan
First published on 21st May 2021
Abstract
We designed a new caging group that can be photoactivated only in the presence of a non-endogenous enzyme when exposed to 405 nm light. Because cells or tissues can be genetically tagged by an exogenously expressed enzyme, this novel method can serve as a strategy for adding targeting abilities to photocaged compounds.
The spatiotemporal control of cellular chemistry using photoreactive molecules offers increasingly essential methods for elucidating the molecular mechanisms that underlie cellular physiology, both in cultured cells and in model organisms.1 Caged compounds are synthetic compounds with a low molecular weight whose biological activities are temporally masked by the covalent modification of a functional group that is critical for their activity; upon photo-irradiation, the temporal mask is removed to restore the original activity. The concept of photocaging was independently reported for the first time by two groups, namely Engels and Schlaeger in 19772 and Kaplan et al. in 1978.3 Since then, the use of caged compounds of biologically relevant molecules has provided an indispensable method for the spatiotemporal control of the cellular physiology, in addition to increasing the number of potential biomedical applications.1,4 The importance of spatiotemporal control in cellular chemistry is recognized, especially in the context of neurotransmission and intracellular signal transduction, since the timings and locations of signaling molecule interactions are strictly regulated in both processes.
Despite the availability of caged compounds exhibiting various chemical and photophysical properties, the lack of spatial resolution and cell-type specificity limit the usefulness of conventional caged compounds for in vivo applications. As conventional caged compounds are not genetically encoded, they cannot be targeted to specific types of cells or tissues of interest. Moreover, it is nearly impossible to precisely focus the activation light on target cells in the highly complex multicellular tissues of model organisms. Recent approaches addressing these issues include caging groups with ligands for organelle localization,5 and the use of modular caging groups that possess chemical handles for connecting additional functionalities, such as cellular targeting abilities.6 Another approach involves the concept of a locked caging group that can be activated by a specific intracellular environment, such as hypoxia7 and DT-diaphorase conditions8 for cancer cell targeting, and thiols for selective protein labeling.9
Inspired by gene-directed enzyme prodrug therapy (GDEPT),10 we herein report the design and synthesis of a new locked photocaging group that is photochemically activated only in the presence of a non-endogenous enzyme. The caging groups are used to prepare caged cyclic nucleotides for proof-of-concept experiments, and the cells of interest are genetically labeled with a non-endogenous enzyme that can restore the photoreactivity of the locked caged compound. Thus, it should be possible to genetically direct the photoreactivities of the new caged compounds to cells or tissues of interest. For the purpose of this study, we design a new caging group, shown in Fig. 1, based on the previously reported (6-bromo-7-hydroxycoumarin-4-yl)methyl (Bhc) caging group, which showed an improved photolysis efficiency under both one- and two-photon excitation conditions.11
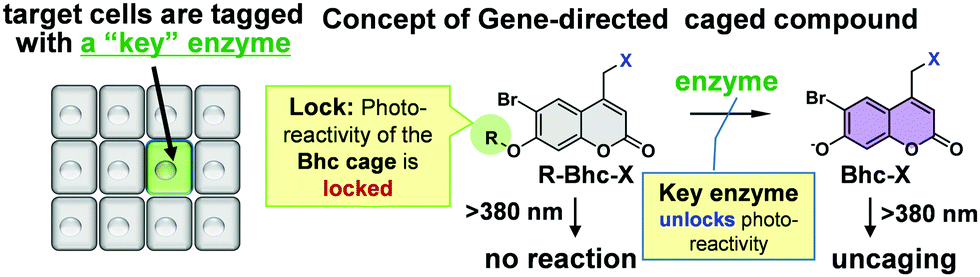 | ||
| Fig. 1 Gene directed targeting of low molecular weight photocaged compounds to the cells of interest. Target cells are genetically tagged with a specific protein. The genetic marker is a key enzyme. The key enzyme unlocks the photo-reactivities of the locked caged compounds. | ||
The Bhc group was selected for use due to several reasons. For example, the electron-withdrawing character of the 6-bromine substituent increases the acidity of the C7-OH group. Since the pKa of the Bhc group is 6.2, approximately 94% of the Bhc group is ionized in aqueous solutions at pH 7.4. The typical absorption maxima for Bhc caged compounds are 325 nm for the protonated form and 375 nm for the ionized form (Fig. 2a), and the molar absorptivity of the ionized Bhc group is approximately 4000 M−1 cm−1 at 405 nm, while that of the protonated Bhc group is nearly zero. When the 7-OH moiety of the Bhc caging group is temporally “locked” by the introduction of an alkyl group, which can later be “unlocked” by a non-endogenous enzyme, the modified Bhc compound (R-Bhc) can be orthogonally photoactivated upon 405 nm irradiation only in the cells expressing the non-endogenous “key” enzyme.
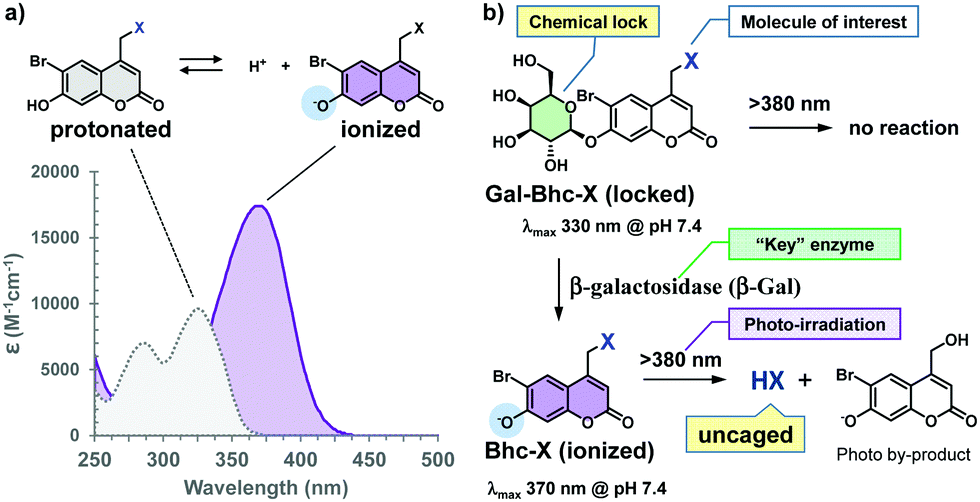 | ||
| Fig. 2 Gal-Bhc group is a new caging group that can be genetically directed. (a) Typical absorption spectra of the Bhc group in aqueous solutions at pH 3 (protonated) and pH 11 (ionized). (b) Gal-Bhc caged compounds should be inert at 380–440 nm irradiation, while unlocked Bhc-caged compounds can release the molecule of interest (uncaged HX). | ||
During this study, we wished to address several points. Firstly, we wanted to confirm that Bhc-cyclic nucleotides can be photolyzed under 405 nm irradiation to produce cyclic nucleotides, while R-Bhc-cyclic nucleotides remain inactive under the same reaction conditions. Secondly, we wanted to determine whether R-Bhc cyclic nucleotides can serve as substrates for their “key” enzymes to yield the corresponding Bhc-cyclic nucleotides while maintaining a satisfactory reactivity. Thirdly, we wished to examine whether the R-Bhc-cyclic nucleotides produce their parent cyclic nucleotides under 405 nm irradiation only inside the cells that express the corresponding “key” enzyme. To address the first two points, we selected the β-galactosyl moiety as a “lock” and β-galactosidase (β-Gal) from Escherichia coli as the “key” (Fig. 2b). The ideal “key” enzyme should not be endogenously expressed, nor should it exhibit endogenous activity. The optimal pH of mammalian β-Gal ranges from 5.5 to 6, whereas commonly used E. coli β-Gal has an optimum pH of 7–7.4. Therefore, the endogenous activity of cytosolic β-Gal is expected to be minimized in mammalian cells, and the β-galactosyl “lock” should be orthogonally unlocked by exogenously expressed E. coli β-Gal.
We therefore designed a new caging group, Gal-Bhc, which contained a β-galactosyl moiety on the 7-OH group of the coumarin ring. Gal-Bhc-cNMPs were synthesized via the corresponding diazomethane derivative, Gal-Bhc-diazo, as shown in Scheme 1. Oxidation of the hydroxymethyl group at the C4 position of 6-bromo-7-methoxymethoxy-4-hydroxymethylcoumarin (MOMBhc-CH2OH, 1)12 was then performed using Dess–Martin periodinane as an oxidizing reagent to give the desired aldehyde, 2, in an excellent yield.
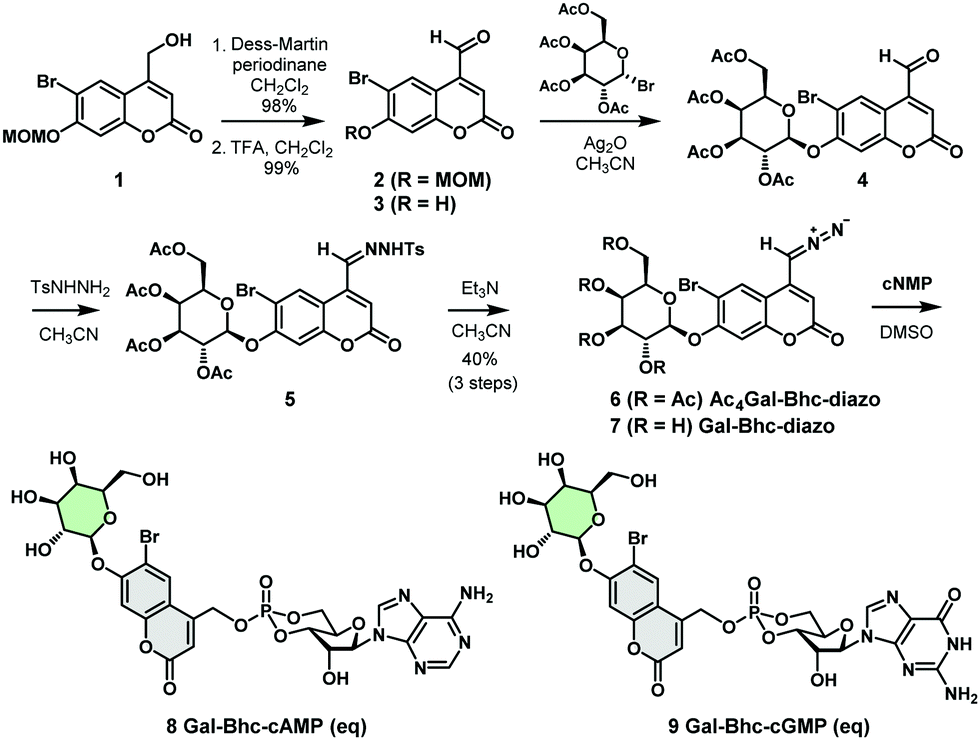 | ||
| Scheme 1 Synthesis of Gal-Bhc-cNMPs. | ||
After deprotection of the MOM group using trifluoroacetic acid (TFA), a quantitative yield of 3 was obtained. The subsequent glycosidation reaction of 3 promoted by silver(I) oxide selectively produced β-galactosyl compound 4 as a single diastereomer, and the condensation of 4 with p-tosylhydrazide gave the corresponding hydrazone 5. Hydrazone 5 was then transformed into AcGal-Bhc-diazo 6 using triethylamine, and treatment of 6 with a basic methanol solution resulted in deprotection of the acetyl groups to give Gal-Bhc-diazo 7. Diazomethyl groups are known to react chemoselectively with phosphate groups in cyclic nucleotides.13 Thus, the reactions of cAMP and cGMP with Gal-Bhc-diazo 7 in DMSO were carried out to obtain Gal-Bhc-cAMP 8 and Gal-Bhc-cGMP 9, respectively. The desired Gal-Bhc-cNMPs (8 and 9) were produced as mixture of their axial and equatorial stereoisomers in terms of their six-membered cyclic phosphate rings. The stabilities of the caged cNMPs were then measured in a simulated physiological solution under dark conditions (Fig. S1, ESI†). It was found that the half-life of Gal-Bhc-cAMP was 209 h for the axial isomer and 146 h for the equatorial isomer, which is similar to those previously reported for Bhc-cNMPs.13
Subsequently, we investigated the photochemical properties of the caged cNMPs. As shown in Fig. S2 (ESI†), the absorption maxima were 374 nm for the Bhc-cNMPs and 327 nm for the Gal-Bhc-cNMPs in a simulated physiological solution (K-MOPS, pH 7.4). The photolysis reactions of the caged cNMPs were therefore performed at 405 nm, and the photolysis mixtures were analyzed periodically and quantified using reversed-phase HPLC. As a result, both Bhc-cNMPs were photolyzed to produce their parent cNMPs, along with Bhc-CH2OH as a photo by-product (Fig. S3, ESI†). The photolytic consumption of the caged cNMPs and the production of cNMPs can be approximated by single exponential equations (Fig. 3), suggesting no unexpected secondary effects that interfere with the photolysis of the Bhc-cNMPs. The disappearance quantum yields (Φdis) of the starting materials were determined to be 0.40 for Bhc-cAMP and 0.43 for Bhc-cGMP under 405 nm irradiation (3.4 mJ s−1). As expected, no photolytic consumption was observed for the Gal-Bhc-cNMPs under 405 nm irradiation (Fig. 3). The photochemical and physical properties of the caged cNMPs tested in this study are summarized in Table S1 (ESI†).
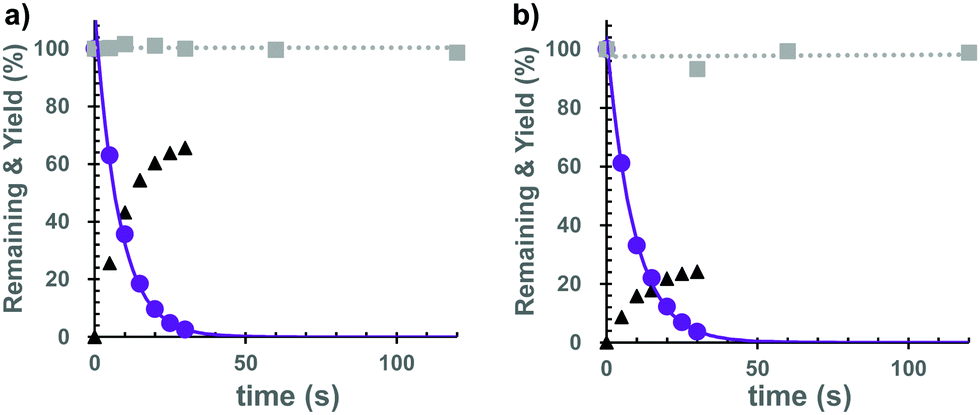 | ||
| Fig. 3 Photolysis of the caged cNMPs. Photolysis reaction time course for the caged cAMPs (a) and cGMPs (b). Samples (10 μM) were irradiated at 405 nm (3.4 mJ s−1) under simulated physiological conditions (pH 7.4). Bhc-cNMP (purple, solid circle), cNMP released from Bhc-cNMP (black, triangle), Gal-Bhc-cNMP (gray, solid square). Solid lines represent the best fits for single exponential decay. Dashed lines represent the results of the linear fitting. | ||
To address the second point mentioned above, we investigated the possibility that Gal-Bhc-cAMP is a substrate of β-Gal that can produce the corresponding Bhc-cAMP. Thus, Gal-Bhc-cAMP was treated with purified β-Gal from E. coli. The progress of the reaction was monitored by absorption changes at wavelengths longer than 380 nm, which were attributed to increasing Bhc-cAMP concentrations (Fig. 4a and Fig. S4, ESI†), and Bhc-cAMP production was observed for both the Gal-Bhc-cAMP isomers. The kinetic parameters of this process were determined by direct fitting of the initial velocity versus the substrate concentration to the Michaelis–Menten equation (Fig. 4b). The Michaelis constant (Km) and apparent reaction rate (kcat) of Gal-Bhc-cAMP were 361 μM and 210 s−1, respectively. The specificity constant (kcat/Km) was determined to be 5.81 × 105 M−1 s−1, which is comparable to those of reported β-Gal substrates, such as ONPG (Km = 120 μM, kcat = 620 s−1, kcat/Km = 5.17 × 106 M−1 s−1)14 and Tb-5 (Km = 81.6 μM, kcat = 2.7 s−1, kcat/Km = 3.31 × 104 M−1 s−1).15 These results indicated that Gal-Bhc-cAMP is a substrate of β-Gal, and that it produces Bhc-cAMP with a comparable reactivity to known substrates that are successfully employed in biochemically useful experiments.
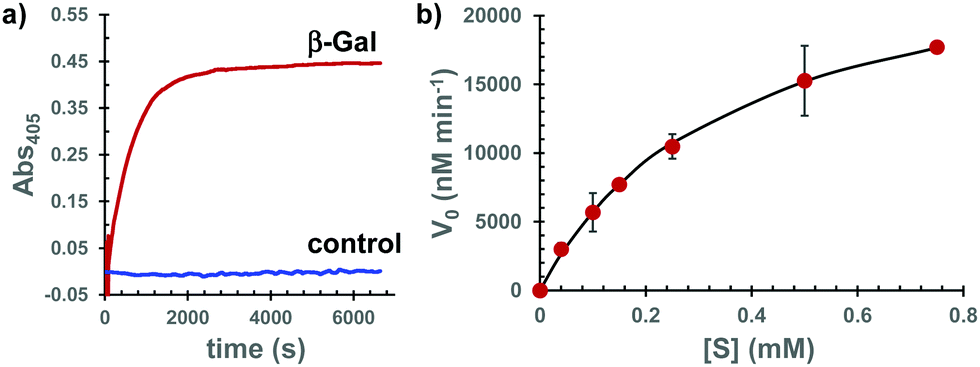 | ||
| Fig. 4 Enzymatic reactions of Gal-Bhc-cAMP. (a) Absorption changes at 405 nm represent the production of Bhc-cAMP at pH 7.4. (b) Plots for the initial velocity (V0) against the substrate concentration for the reaction of Gal-Bhc-cAMP with E. coli β-Gal (2.1 nM). Data points represent the averages of duplicated experiments ± the standard deviation (SD) of the mean. Solid line is the best fit for the Michaelis–Menten equation. | ||
Subsequently, we examined whether the transiently expressed β-Gal could produce a detectable amount of Bhc-cAMP in cultured mammalian cells. For this purpose, HEK 293T cells were transfected with a plasmid encoding E. coli β-Gal. Gal-Bhc-cAMP (10 μM) was incubated with cell lysates at 37 °C for 2 h, and the obtained mixtures were analyzed by reversed-phase HPLC. The HPLC traces shown in Fig. S5 (ESI†) indicate that Bhc-cAMP production was detected within 2 h of incubation with the β-Gal-expressing cell lysates (Fig. S5a and b, ESI†). Following treatment with the lysates for 3 h, Gal-Bhc-cAMP was irradiated with 405 nm light (3.4 mJ s−1) for 20 s, and the quantitative production of cAMP was observed from the β-Gal-expressing cell lysates (Fig. S5c, ESI†). However, almost no detectable production of Bhc-cAMP was observed in the MOCK-treated cells (Fig. S5d, ESI†). Similar results were obtained for Gal-Bhc-cGMP (Fig. S5e–h, ESI†), thereby confirming that the Gal-Bhc-cNMPs are selectively uncaged to produce their parent cNMPs only in the presence of their key enzyme, β-Gal.
Based on these results, we applied the Gal-Bhc-cNMPs to live HeLa cells transiently expressing E. coli β-Gal (lacZ) for addressing the third point mentioned above. Changes in intracellular cAMP concentrations were monitored using the genetically encoded fluorescent indicator, R-FlincA.16 Thus, the HeLa cells were co-transfected with two plasmid DNA vectors (lacZ and R-FlincA), and following incubation with 10 μM Gal-Bhc-cAMP, the cells were exposed to 405 nm light under a fluorescent microscope. Changes in the fluorescence intensity of R-FlincA before and after light exposure were recorded (Fig. S6, ESI†). Since not all cells were transfected with both lacZ and R-FlincA, two types of cells were expected to be present in the same dish: β-Gal(+) cells expressing both lacZ and R-FlincA, and β-Gal(−) cells expressing only R-FlincA. β-Gal(+) cells responded to both forskolin (an adenylyl cyclase activator) and FDG-C12 (a membrane-permeable fluorescent β-Gal indicator), whereas β-Gal(−) cells responded to forskolin but not to FDG-C12. Representative time courses for the fluorescence changes observed for R-FlincA in the β-Gal(+) and β-Gal(−)cells are shown in Fig. 5 and Fig. S7 (ESI†). Following 405 nm irradiation, a slight increase in the fluorescence intensity was observed for the β-Gal(+) cells (Fig. 5a and Fig. S7b, ESI†), while no significant change was detected for the β-Gal(−)cells (Fig. 5b and Fig. S7d, ESI†). The changes in the R-FlincA fluorescence intensity (ΔF/F0) in the β-Gal(+) and β-Gal(−) cells (n = 46 and 39, respectively) were then quantified (Fig. 5c). A statistically significant increase was observed for the β-Gal(+) cells compared to the β-Gal(−) cells. Because Bhc-cAMP exhibits a low membrane permeability,13 the cell-to-cell transfer of the produced Bhc-cAMP and cAMP itself should be negligible throughout the experiments. This was further confirmed by the fact that no significant difference was observed between the β-Gal(−) cells and the control cells that were transfected with R-FlincA alone (n = 83). Based on these results, we concluded that Gal-Bhc-cAMP penetrates live mammalian cells and produces cAMP upon 405 nm irradiation only in cells that express the “key” enzyme, E. coli β-Gal.
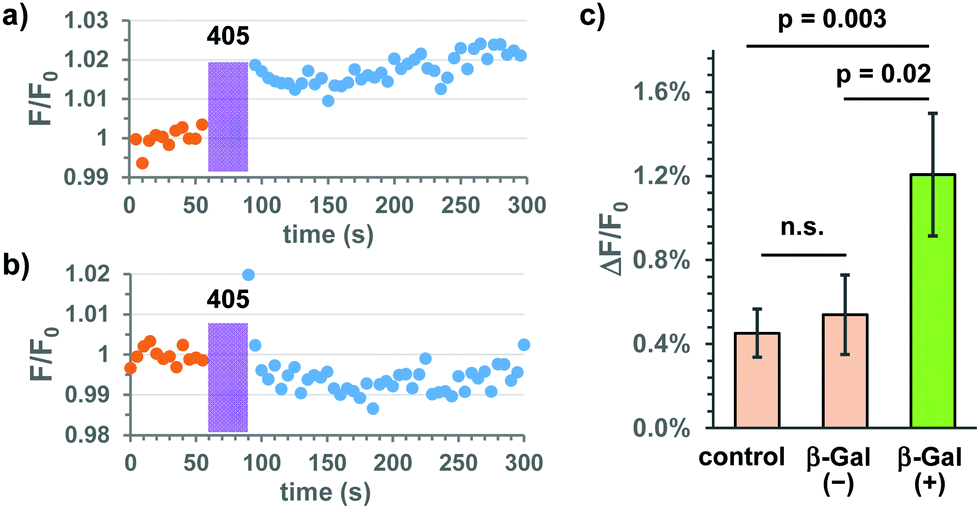 | ||
| Fig. 5 Photo-mediated selective cAMP production in cultured mammalian cells transiently expressing E. coli β-Gal. HeLa cells were co-transfected with plasmids coding for E. coli β-Gal and R-FlincA. All cells were incubated with Gal-Bhc-cAMP (10 μM) for 24 h and exposed to 405 nm light (3.4 mJ s−1) for 30 s. Traces showing the fluorescence changes of R-FlincA in (a) β-Gal(+) and (b) β-Gal(−) cells. (c) Quantification of the changes in fluorescence intensity (ΔF/F0) before and after 405 nm irradiation. F0 denotes the initial fluorescence intensity before photo-irradiation, while ΔF denotes the difference obtained by subtracting the intensity before light irradiation from the intensity after light irradiation. Data are means ± standard errors (SE) for control cells, β-Gal(−) cells, and β-Gal(+) cells. Student T-tests were used for the determination of a statistical significance (n.s. denotes not significant). | ||
In conclusion, we designed a new gene-directed caging group, namely the Gal-Bhc group, which can be photoactivated only in the presence of its “key” enzyme, E. coli β-Gal, when exposed to 405 nm light. A precursor molecule, Gal-Bhc-diazo, was synthesized and used for the preparation of Gal-Bhc-caged cyclic nucleotides, i.e., Gal-Bhc-cAMP and Gal-Bhc-cGMP. It was found that Gal-Bhc-diazo reacted chemoselectively with the phosphates of the cNMPs, suggesting that other nucleotides can also be transformed into Gal-Bhc caged compounds without the requirement for protecting groups. As cells or tissues can be genetically tagged with the key enzyme without an apparent significant effect on the cells of interest, “locked” caged compounds, such as Gal-Bhc caged compounds, have the potential for use as gene-directed caged compounds that are genetically targeted to the cells and tissues of interest. This would increase the utility of currently available caged compounds in biomedical applications.
With regards to Gal-Bhc caged cyclic nucleotides, the increase in cAMP concentration in β-Gal expressing cells that we observed was less than expected. We hypothesized that this was due to reduced membrane permeability of these compounds. Its estimated ClogP value is −1.7. A modification of the Gal-Bhc group with a hydrophobic substituent would enhance membrane permeability. Further research to pursue this line of inquiry is underway.
This work was supported by JSPS KAKENHI grant number 17H05770 (TF and KH), “Advanced Bioimaging,” and JP19H05778 (TF), “MolMovies.” We thank Dr Takuya Tamura for his helpful comments and discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. Ankenbruck, T. Courtney, Y. Naro and A. Deiters, Angew. Chem., Int. Ed., 2018, 57, 2768 CrossRef CAS PubMed.
- J. Engels and E. J. Schlaeger, J. Med. Chem., 1977, 20, 907 CrossRef CAS PubMed.
- J. H. Kaplan, B. Forbush, 3rd and J. F. Hoffman, Biochemistry, 1978, 17, 1929 CrossRef CAS PubMed.
- (a) P. Klan, T. Solomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef CAS PubMed; (b) G. Bort, T. Gallavardin, D. Ogden and P. I. Dalko, Angew. Chem., Int. Ed., 2013, 52, 4526 CrossRef CAS PubMed; (c) M. Abe, Y. Chitose, S. Jakkampudi, P. T. T. Thuy, Q. Lin, B. T. Van, A. Yamada, R. Oyama, M. Sasaki and C. Katan, Synthesis, 2017, 3337 CAS; (d) G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900 CrossRef CAS PubMed; (e) Y. Hou, Z. Zhou, K. Huang, H. Yang and G. Han, ChemPhotoChem, 2018, 2, 1005 CrossRef CAS; (f) M. Karimi, P. Sahandi Zangabad, S. Baghaee-Ravari, M. Ghazadeh, H. Mirshekari and M. R. Hamblin, J. Am. Chem. Soc., 2017, 139, 4584 CrossRef CAS PubMed; (g) Y. Bao, L. Y. Zhu, Q. N. Lin and H. Tian, Adv. Mater., 2015, 27, 1647 CrossRef PubMed.
- (a) T. Horinouchi, H. Nakagawa, T. Suzuki, K. Fukuhara and N. Miyata, Bioorg. Med. Chem. Lett., 2011, 21, 2000 CrossRef CAS PubMed; (b) A. Leonidova, V. Pierroz, R. Rubbiani, Y. J. Lan, A. G. Schmitz, A. Kaech, R. K. O. Sigel, S. Ferrari and G. Gasser, Chem. Sci., 2014, 5, 4044 RSC; (c) A. Nadler, D. A. Yushchenko, R. Müller, F. Stein, S. Feng, C. Mulle, M. Carta and C. Schultz, Nat. Commun., 2015, 6, 10056 CrossRef CAS PubMed; (d) S. H. Feng, T. Harayama, S. Montessuit, F. P. A. David, N. Winssinger, J. C. Martinou and H. Riezman, eLife, 2018, 7, e34555 CrossRef PubMed; (e) N. Wagner, M. Stephan, D. Hoglinger and A. Nadler, Angew. Chem., Int. Ed., 2018, 57, 13339 CrossRef CAS PubMed; (f) S. Feng, T. Harayama, D. Chang, J. T. Hannich, N. Winssinger and H. Riezman, Chem. Sci., 2019, 10, 2253 RSC.
- (a) T. Furuta, K. Manabe, A. Teraoka, K. Murakoshi, A. Ohtsubo and A. Suzuki, Org. Lett., 2012, 14, 6182 CrossRef CAS PubMed; (b) A. Teraoka, K. Murakoshi, K. Fukamauchi, A. Z. Suzuki, S. Watanabe and T. Furuta, Chem. Commun., 2014, 50, 664 RSC; (c) A. Z. Suzuki, R. Sekine, S. Takeda, R. Aikawa, Y. Shiraishi, T. Hamaguchi, H. Okuno, H. Tamamura and T. Furuta, Chem. Commun., 2019, 55, 451 RSC.
- (a) W. P. Feng, C. Y. Gao, W. Liu, H. H. Ren, C. Wang, K. Ge, S. H. Li, G. Q. Zhou, H. Y. Li, S. X. Wang, G. Jia, Z. H. Li and J. C. Zhang, Chem. Commun., 2016, 52, 9434 RSC; (b) Q. N. Lin, C. Y. Bao, Y. L. Yang, Q. N. Liang, D. S. Zhang, S. Y. Cheng and L. Y. Zhu, Adv. Mater., 2013, 25, 1981 CrossRef CAS PubMed; (c) M. Gangopadhyay, R. Mengji, A. Paul, Y. Venkatesh, V. Vangala, A. Jana and N. D. P. Singh, Chem. Commun., 2017, 53, 9109 RSC.
- Z. Chen, B. Li, X. Xie, F. Zeng and S. Wu, J. Mater. Chem. B, 2018, 6, 2547 RSC.
- (a) Q. Lin, C. Bao, S. Cheng, Y. Yang, W. Ji and L. Zhu, J. Am. Chem. Soc., 2012, 134, 5052 CrossRef CAS PubMed; (b) Q. Lin, Z. Du, Y. Yang, Q. Fang, C. Bao, Y. Yang and L. Zhu, Chem. – Eur. J., 2014, 20, 16314 CrossRef CAS PubMed.
- Z. Ram, K. W. Culver, S. Walbridge, R. M. Blaese and E. H. Oldfield, Cancer Res., 1993, 53, 83 CAS.
- (a) T. Furuta, S. S. H. Wang, J. L. Dantzker, T. M. Dore, W. J. Bybee, E. M. Callaway, W. Denk and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1193 CrossRef CAS PubMed; (b) T. Furuta, J. Synth. Org. Chem., Jpn., 2012, 70, 1164 CrossRef CAS.
- A. Z. Suzuki, T. Watanabe, M. Kawamoto, K. Nishiyama, H. Yamashita, M. Ishii, M. Iwamura and T. Furuta, Org. Lett., 2003, 5, 4867 CrossRef CAS PubMed.
- T. Furuta, H. Takeuchi, M. Isozaki, Y. Takahashi, M. Kanehara, M. Sugimoto, T. Watanabe, K. Noguchi, T. M. Dore, T. Kurahashi, M. Iwamura and R. Y. Tsien, ChemBioChem, 2004, 5, 1119 CrossRef CAS PubMed.
- D. H. Juers, B. Rob, M. L. Dugdale, N. Rahimzadeh, C. Giang, M. Lee, B. W. Matthews and R. E. Huber, Protein Sci., 2009, 18, 1281 CrossRef CAS PubMed.
- K. Hanaoka, K. Kikuchi, T. Terai, T. Komatsu and T. Nagano, Chem. – Eur. J., 2008, 14, 987 CrossRef CAS PubMed.
- Y. Ohta, T. Furuta, T. Nagai and K. Horikawa, Sci. Rep., 2018, 8, 1866 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc01405f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |