
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycodendritic structures as DC-SIGN binders to inhibit viral infections
Javier
Ramos-Soriano
and
Javier
Rojo
 *
*
Glycosystems Laboratory, Instituto de Investigaciones Químicas (IIQ), Centro de Investigaciones Científicas Isla de La Cartuja, CSIC and Universidad de Sevilla, Américo Vespucio, 49, 41092 Sevilla, Spain. E-mail: javier.rojo@iiq.csic.es
First published on 30th April 2021
Abstract
DC-SIGN, a lectin discovered two decades ago, plays a relevant role in innate immunity. Since its discovery, it has turned out to be a target for developing antiviral drugs based on carbohydrates due to its participation in the infection process of several pathogens. A plethora of carbohydrate multivalent systems using different scaffolds have been described to achieve this goal. Our group has made significant contributions to this field, which are revised herein.
 Javier Ramos-Soriano | Javier Ramos-Soriano received his Doctor of Chemistry degree in 2017. Between 2017 and 2019, he pursued postdoctoral research at Universidad Complutense de Madrid (Spain) to work on the synthesis of glyconanomaterials as antiviral agents. In 2019, he gained an MSCA Fellowship in the Carmen Galán group (University of Bristol) working on bio-inspired glycan-nanoprobes as antimicrobial, anticancer, and antiviral pro-drugs. Recently, he joined the Spanish National Research Council (CSIC) as a Talent Hub Postdoctoral Fellow. His present research interests are focused on the development of glycodendritic systems as tools to understand and intervene in biological processes related to pathogen infections and immune systems. |
 Javier Rojo | Dr Javier Rojo obtained his PhD in Chemistry (1995) from the Autónoma University of Madrid (UAM) in Spain, under the supervision of Prof. Juan Carlos Carretero. He was a postdoctoral fellow with Prof. Jean-Marie Lehn at the University Louis Pasteur in Strasbourg (1995–1998) and with Prof. Larry W. McLaughlin at Boston College in the USA (1998–1999). In January 2000, he joined the Carbohydrate Group at the Institute of Chemical Research of CSIC as an Associate Researcher. He is currently a Research Scientist at CSIC in Seville. His current research interests include carbohydrates, dendrimers, bioconjugates, and molecular recognition. |
Introduction
In 1992, the lectin CD209 was identified during a broad screening of potential membrane receptors for gp120, the envelope glycoprotein of HIV-1.1 This discovery remained unnoticed for almost eight years until the work of van Kooyk and Littman brought it to light,2 and this seminal paper made a huge impact in the scientific community. For the first time, it was shown how a receptor, CD209, renamed DC-SIGN (Dendritic Cells Specific Intercellular adhesion molecule 3 Grabbing Non-integrin),3 facilitated the trans-infection of T-cells by HIV-1. Since then, the intensive work developed by several groups around the world has been devoted to unravelling the role that this lectin could play in pathogen infection.4–8DC-SIGN is a C-type lectin with a Carbohydrate Recognition Domain (CRD) at the C-terminus exposed to the cytoplasm, which is capable of interacting efficiently with sugars.9,10 To gain better insight into this recognition process, the types of oligosaccharides that can be ligands for this lectin have been explored. The branched high mannose oligosaccharide (Man9GlcNAc2) was identified as one of the natural ligands for this receptor, found in several viral envelope glycoproteins such as the gp120 of HIV11 or the GP1 of Ebola.12 Apart from high mannose, other oligosaccharides, mainly fucosylated, such as Lex, Ley, and blood antigens, are also recognised by DC-SIGN.13 There are very few approaches where other types of molecules that are not carbohydrates have been explored as antagonists for DC-SIGN.14 However, the real application of these types of compounds has not been explored, probably due to the lack of selectivity.
DC-SIGN is mainly expressed at the surface of immature dendritic cells as homotetramers. This lectin is clustered at specific sites of the cell membrane, creating lectin patches that undoubtedly have a pivotal role in pathogen recognition, uptake and intracellular signalling.15 Homotetrameric DC-SIGN has a square-like arrangement of its oligomannose binding sites that are separated by around 40 Å (Fig. 1).16 Due to the low affinity that is usually present in carbohydrate–protein interactions,17 this special disposition of tetramer clustering creates an ideal multivalent surface to capture pathogens through multivalent interactions with the highly glycosylated pathogen envelope glycoproteins.
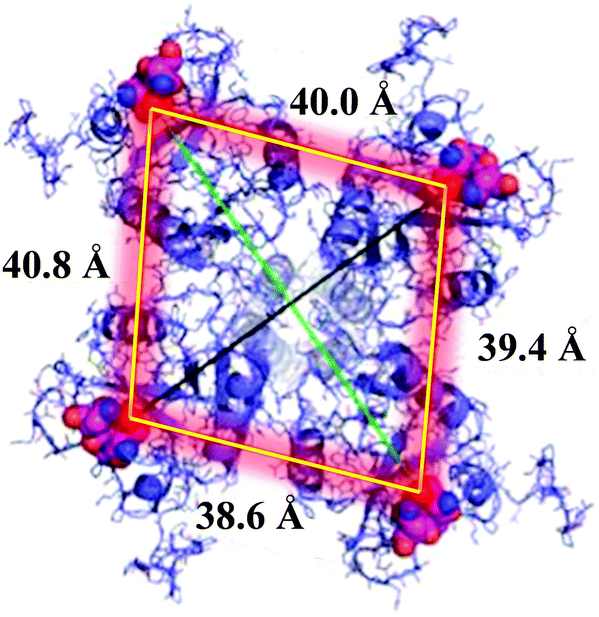 | ||
| Fig. 1 The DC-SIGN homotetramer and the distance between the CRDs. Image adapted from Glycopedia. | ||
In the particular case of HIV-1, DC-SIGN is the gate for virus entrance, facilitating the infection of cells. For this reason, the design of inhibitors capable of competing with this virus, blocking the receptor, has been a very attractive strategy for the design of antiviral compounds. The most affordable approach requires the design of carbohydrate multivalent systems to increase the binding affinity. Therefore, multivalent binding could involve the simultaneous interaction of all four binding sites of DC-SIGN. If the multivalent system is big enough, the system could interact simultaneously with more than one receptor at the cell surface, covering a large area and blocking several receptors even without interacting physically with them. Moreover, the rebinding of carbohydrate ligands to the receptor is also a typical effect found in multivalent interactions. The chance to find a ligand close to the receptor in an appropriate disposition increases with the valency and provides an increment in the binding affinity typically found for these systems. In this context, these carbohydrate multivalent systems could inhibit the infection process mediated by this receptor. Toward this aim, a plethora of different multivalent scaffolds are available to prepare this type of compound such as dendrimers,18,19 gold nanoparticles,20 liposomes,21 polymers,22 proteins,23 among other scaffolds, have been explored.
In this article, we summarize the contribution of our group to this area, as well as relevant contributions provided by other research groups. This covers the research of the last 20 years, from simple compounds to very sophisticated ones with antiviral activities in the μM to pM range.
Mannosylated glycodendrimers
Our contribution to this field dates back to the end of 2002, just a couple of years after the paramount discovery of the role that DC-SIGN plays in the T-cell trans-infection by HIV-1. With all the information available concerning carbohydrate–protein interactions, it was clear that systems with several copies of sugars should be required to achieve a reasonable affinity to interact efficiently with DC-SIGN. As a proof of concept, we designed a very affordable system based on a commercially available hyperbranched polymer with a low polydispersity degree (PD < 1.4), the Boltorn polyester.24 This is a very cheap compound that is available in different generations (G) such as BH20 (2G), BH30 (3G) and BH40 (4G) with 16, 32 and 48 hydroxyl groups at the surface, respectively. As a sugar, we decided to use the simplest ligand for DC-SIGN, a mannose monosaccharide. Mannoses were conjugated to the scaffold via the formation of amides using the unprotected sugars. Carboxylic acids were introduced into the Boltorn surface using succinic anhydride to obtain compound 1, followed by coupling with mannose 2 provided with an amino short spacer at the anomeric position (Fig. 2). By this means, we prepared the 2nd and the 3rd generations of Boltorn glycopolymers with an average of 16 and 30 mannoses, respectively.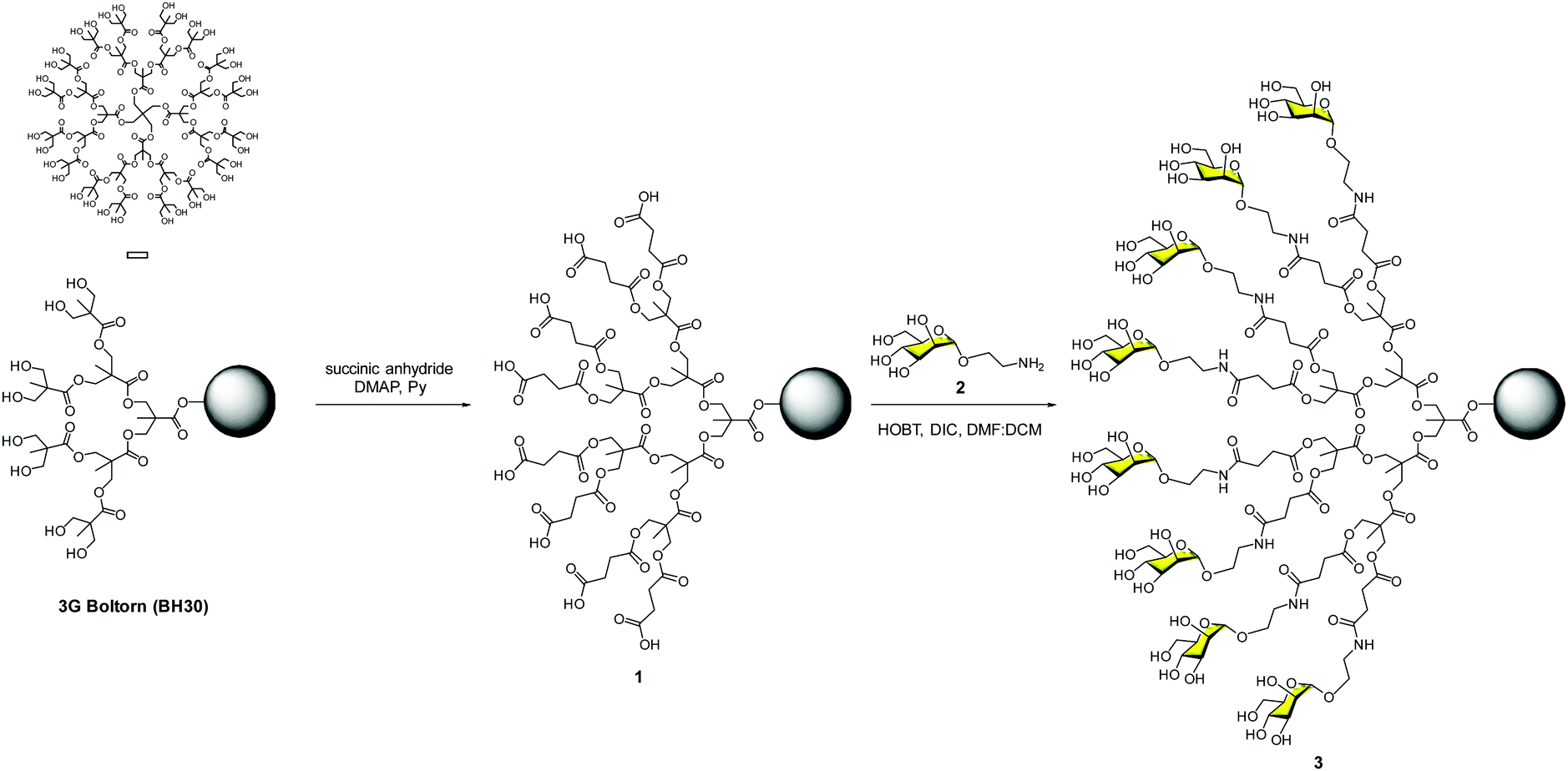 | ||
| Fig. 2 Synthesis of the 3G Boltorn-type glycodendrimer 3 with 32 mannoses. | ||
These glycopolymers were first tested in a turbidimetry assay with Lens culinaris, a plant lectin that recognises mannoses. These experiments demonstrated the ability of the 3G Boltorn glycopolymer 3 to interact in a multivalent way with this lectin producing the precipitation of complex clusters. With these preliminary results, we moved to cell assays to test the ability of these glycopolymers to inhibit the infection of Jurkat cells in a DC-SIGN-dependent manner using pseudotyped Ebola virus particles.25 We demonstrated that glycopolymer 3 inhibited this infection in the low μM range (IC50 = 0.3 μM). Moreover, this glycopolymer inhibited the trans-infection of K562 cells very efficiently. Taking, the IC50 of monovalent mannose (1.27 mM) into account, these data highlighted the multivalent effect. Glycopolymer 3 was two orders of magnitude more potent per mannosyl unit when compared to the simple mannose. We also demonstrated that glycopolymer 3 was able to inhibit the interaction of HIV-1 gp120 with DC-SIGN in micromolar concentrations (IC50 = 50 μM) using a Surface Plasmon Resonance (SPR) biosensor.26 These results opened the door to exploring these carbohydrate multivalent systems as antiviral drugs.27
In 2012, we reported the synthesis of a monodisperse dendron with nine mannoses 4 (Fig. 3) to analyse the interaction with DC-SIGN at the surface of dendritic cells.28 We used BODIPY for fluorescence labelling at the focal position of the dendron (compound 5, Fig. 3) and evaluated the interaction and the uptake by flow cytometry and confocal microscopy. The results revealed that this relatively simple glycodendron interacted efficiently with the cell surface and was internalised via a receptor-dependent mechanism at physiological temperature. The interaction did not promote cell maturation or cytokine expression, at least at the concentration tested. The glycodendron 5 was stored inside the cell within late endosomes. This process was inhibited when a competitor such as mannan or a specific CRD DC-SIGN antibody was applied.
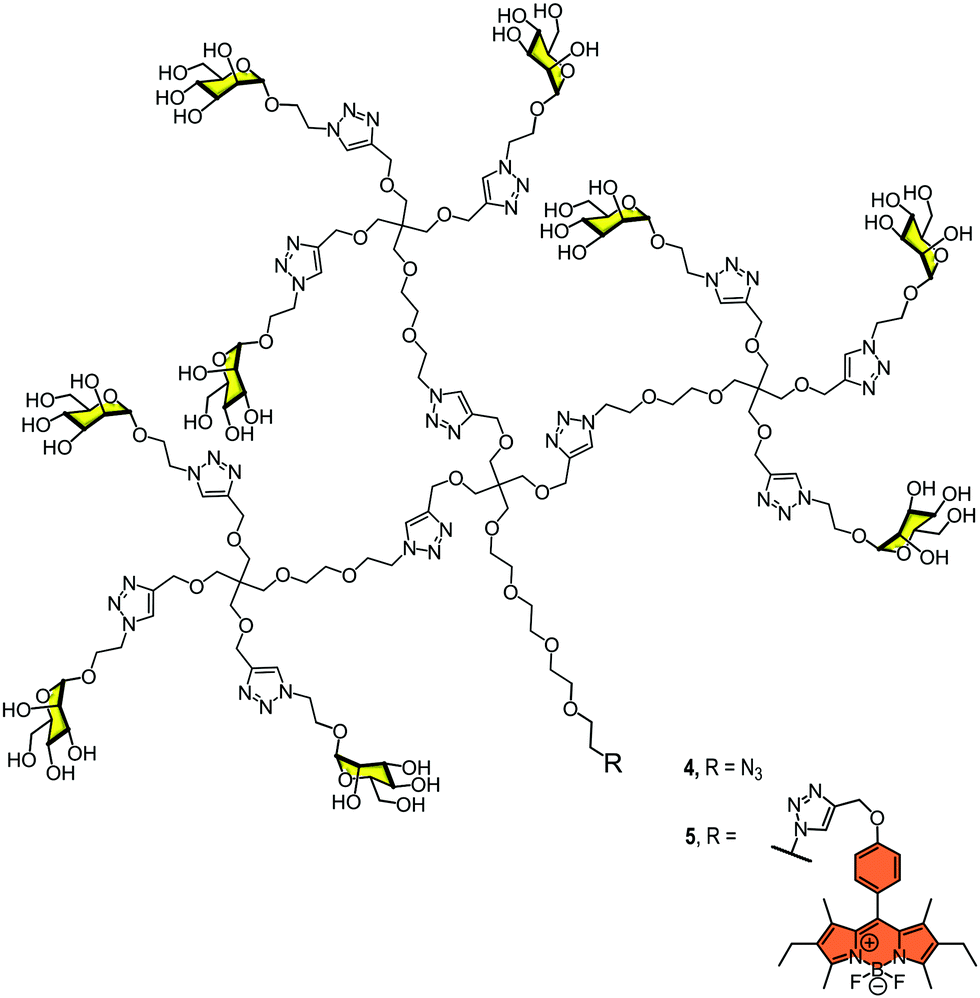 | ||
| Fig. 3 Glycodendron of nine mannoses unlabelled (4) and labelled (5) with the BODIPY probe. | ||
These promising preliminary results encouraged us to improve the inhibitory ability of this multivalent system by employing different ligands. In this context, our research group and the Bernardi group carried out multivalent systems decorated with pseudomannobiose 629 and pseudomannotriose 830 sugars (Fig. 4). They are mimetics of the mannose disaccharide Manα1,2Man and the linear trisaccharide Manα1,2Manα1,2Man, respectively, as ligands for DC-SIGN. These new ligands presented some interesting features, being better binders to DC-SIGN than the simple mannose. Moreover, they presented strong stability against enzymatic degradation by glycosidases in comparison to their corresponding natural carbohydrates.31
 | ||
| Fig. 4 General structures of pseudomannobiose (6–7), pseudomannotriose (8–9) and modified pseudomannobiose (10) glycomimetics. | ||
Firstly, we carried out the preparation of the Boltorn scaffolds using a stepwise synthesis instead of using the polydisperse commercial one. For this purpose, a sequential synthesis using the pentaerythritol as the core and the subsequent coupling with a protected anhydride of 2,2-bis(hydroxymethyl)propionic acid (bis-MPA) 12 were carried out (Fig. 5A). After the last deprotection step, the reaction with the succinyl derivative 14 provided the functionalization of the surface with carboxylic acids. The conjugation with the glycomimetics 6 and 8 yielded the corresponding glycodendrimers with 32 pseudodisaccharides 15a and pseudotrisaccharides 15b (Fig. 5A).32
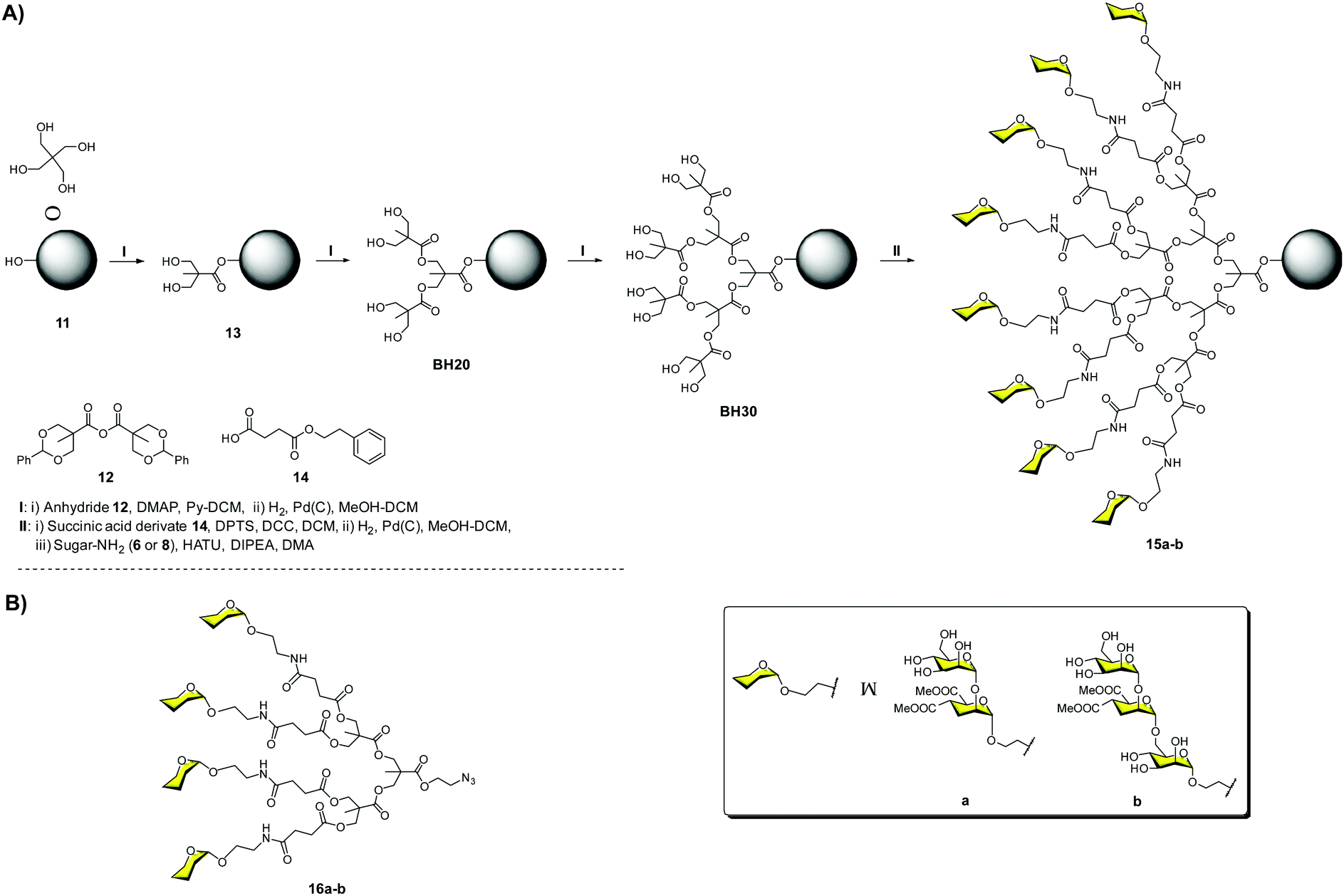 | ||
| Fig. 5 Stepwise synthesis of Boltorn-type glycodendrimers 15a–b (A) and general structures of tetravalent dendrons 16a–b (B). | ||
The activity of these compounds was evaluated in the same Ebola infection model described previously. The results were very promising, achieving inhibitory activities in the nanomolar range (IC50 = 17 and 27 nM for 15b and 15a, respectively), confirming the ability of these compounds to block the infection. Surprisingly, no significant differences were found for the pseudomannobioside and pseudomannotrioside 3G glycodendrimers in terms of activity. However, tetravalent dendron 16b (one of the arms of the glycodendrimers) decorated with the pseudotrisaccharide (IC50 = 0.1 μM) was one order of magnitude more potent than the corresponding pseudodisaccharide dendron 16a (IC50 = 1.4 μM) (Fig. 5B). Similar results were found in the trans-infection experiment, obtaining IC50 in the same range.
Dendrons 16a–b were also evaluated in an HIV infection model using B-THP-1 cells expressing DC-SIGN and several HIV strains that were lab adapted and primarily isolated from infected patients.33 In these assays, the trans-infection of CD4+ cells was inhibited with an IC50 of 5 μM for the tetravalent pseudomannotriose dendron 16b, independently of the viral strain, which was a remarkable feat, being the inhibition higher than 90% when a concentration of 1 μM was used. This compound was also tested using an explant cervix tissue.34 This very interesting model represents an approach to the site where HIV infection usually occurs after sexual intercourse. The tetravalent pseudotrisaccharide dendron 16b demonstrated a strong inhibitory activity, blocking around 80–90% of the HIV-1 viral infection. This tetravalent dendron induced the production of β-chemokines responsible for interacting with the CCR5 co-receptor of the HIV-1 R5 strain, reducing the replication of this virus. In addition, compound 16b showed low toxicity and presented a good penetration capacity in the epithelium tissue due to its small size. For all these reasons, it was considered as a potential lead for the development of microbicides for topical use before high-risk sexual encounters.
The very good results observed for this small compound prompted us to explore the combination of small and more accessible simple scaffolds with a low-medium valency.35 Initially, a series of glycomimetics was synthesized (Fig. 4) and tested using SPR biosensors. Among them, compound 10, an evolution of the former pseudodisaccharide 7 where the methyl esters were replaced by amides with an aromatic ring, showed the most promising behaviour. Glycodendrimers with 4, 6, 9, 12, and 18 copies of pseudodisaccharide 7, pseudotrisaccharide 9, and the new modified pseudodisaccharide 10 were prepared (Fig. 6). The sugars were conjugated to the dendrimeric scaffolds via a Cu(I)-catalysed alkyne azide cycloaddition (CuAAC) click chemistry reaction. For this coupling, the scaffolds were decorated with alkyne groups and the sugars presented an azide at the end of the short spacer at the anomeric position. The dendrimeric scaffolds were small commercially available molecules with 3, 4, or 6 functional points. The valency could be increased by coupling trivalent glycodendrons 17 on these scaffolds to achieve higher valencies (9, 12 and 18) using a convergent strategy.
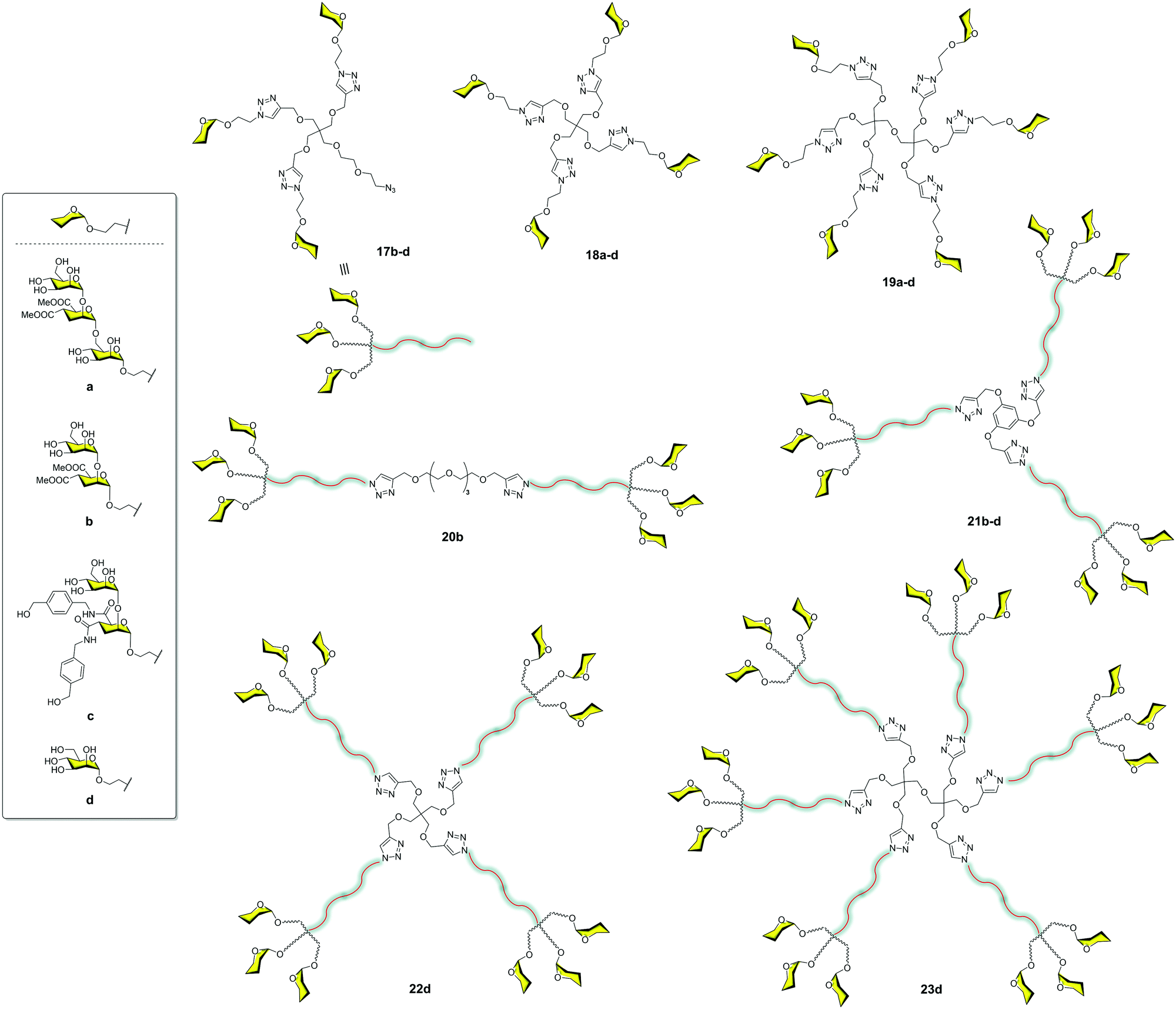 | ||
| Fig. 6 General structures of glycodendron (17) and glycodendrimers (18–23). | ||
The activity of these compounds 17–23 was tested in competition experiments using an SPR biosensor with immobilised Man–BSA on the chip and DC-SIGN. The results showed an increase in the affinity of these glycodendrons with the increase of valency. In particular, glycodendrons functionalized with the new pseudodisaccharide 10 were the most interesting systems. The corresponding tetra and hexadendrimers of this pseudodisaccharide 18c and 19c, respectively, were evaluated to inhibit the HIV-1 trans-infection of T-cells. The hexavalent dendrimer 19c showed an IC50 of 1 μM, being that the infection process was totally inhibited at 10 μM, a remarkable activity that revealed the potential of this compound. The antiviral activity was also evaluated against a different pathogen that uses DC-SIGN for infection such as the Dengue virus. The same glycodendrimer was able to inhibit the infection of Raji-DC-SIGN cells in a dose-dependent manner with an IC50 of 5.9 μM.
Moravcová et al.36 designed C-glycomimetics of mannose and L-fucose as ligands for DC-SIGN that were resistant to enzymatic degradation by glycosidases. These compounds were conjugated to different scaffolds via a CuAAC click chemistry reaction (Fig. 7). The glycodendrimers 24a–c, 25b–c, 26b–c, 27a–c and 28a–c were evaluated by SPR competition assays using a Man–BSA-functionalized chip and the soluble tetravalent DC-SIGN. IC50 values in the micromolar range were found for the dodecavalent C-mannose 28a and C-fucose 28b glycodendrimers. These compounds showed similar activity to that found for the corresponding O-partners but with greater stability. This work demonstrated that these glycomimetics could be a good alternative to natural sugars in order to use more stable compounds.
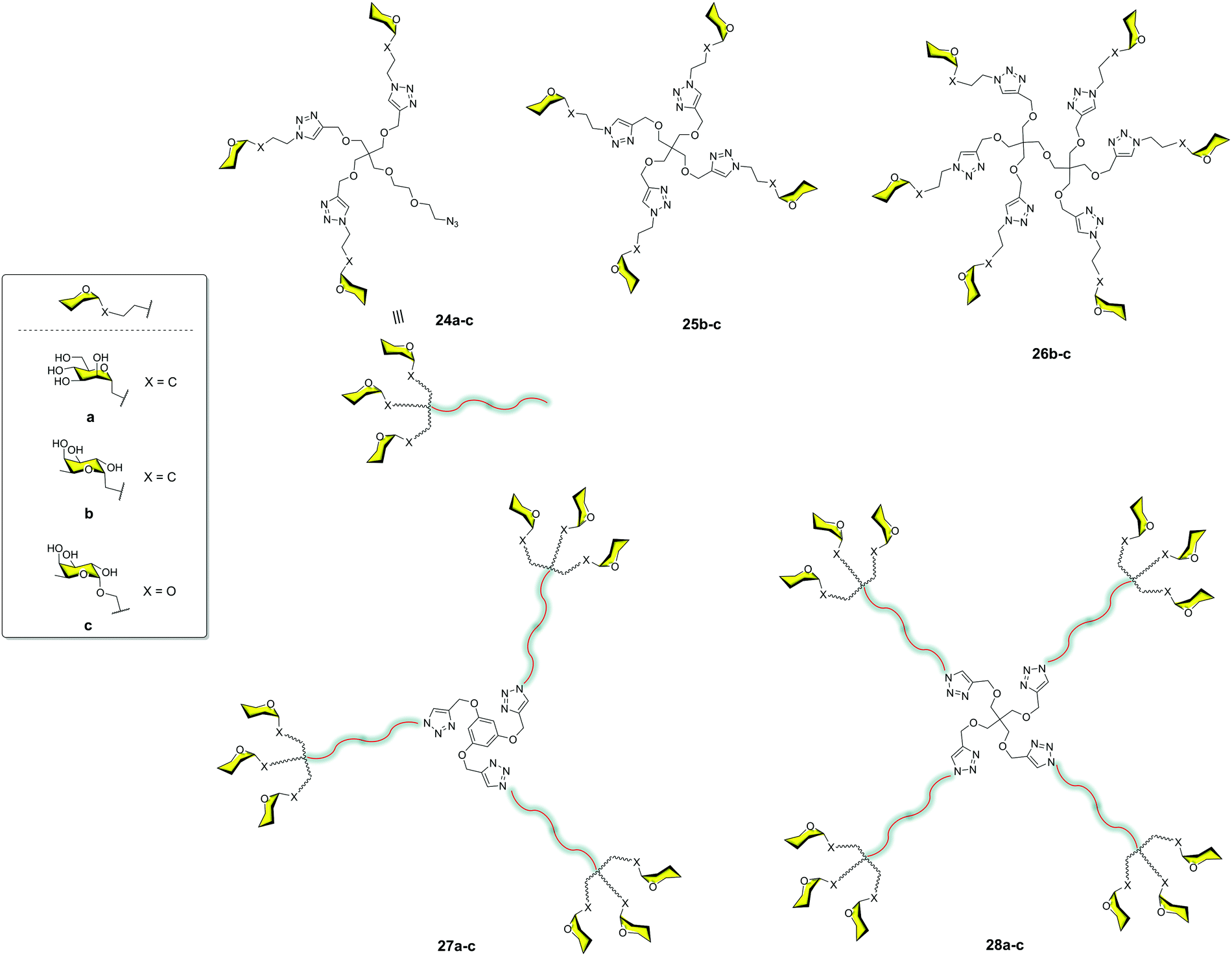 | ||
| Fig. 7 General structures of glycodendron (24) and glycodendrimers (25–28) decorated with C- and O-glycosides. | ||
All of these contributions summarised here represent a strategy focused on relatively simple and accessible scaffolds. The role of the scaffold was exclusively to allow a multivalent presentation of the ligands. Moreover, higher affinities were obtained by increasing the valency without focusing on the design of scaffolds to allow an adequate distance between ligands to simultaneously reach at least two CRDs of the DC-SIGN tetramer. In this context, the van Kooyk group reported the preparation of glycodendrimers based on Lewis-type antigens (LeX, Lea, Leb) and the commercially available dendrimer PAMAM;37 they used the generations from 3 to 5. The dendrimers were functionalized with a similar number of sugars, around 16. In this way, the distances between sugars were different due to the size of the dendrimer, which was between 1.8 nm and 2.7 nm depending on the generation. The larger compound was the better competitor to inhibit the binding between the gp120 of HIV and DC-SIGN. The results demonstrated that the distance, more than the number of sugars, was the most relevant issue. This G5 LeX–PAMAM dendrimer was able to completely inhibit the trans-infection of CD4+ cells after 48 hours. In addition, this compound showed selectivity for DC-SIGN versus Langerin, a key point for the design of HIV-1 inhibitors.
Bernardi and co-workers38 described a multivalent scaffold based on a semi-rigid rod whose ends were functionalized with glycodendrons (Fig. 8A). The distance between both ends was adequate to simultaneously reach two CRDs on a DC-SIGN tetramer as shown by molecular dynamics simulations. Compound 29 with six pseudodisaccharides (Fig. 8A) inhibited the trans-infection of CD4+ T-lymphocytes by HIV-1 with an IC50 in the nM range. This was the first example that demonstrated the relevance of a careful design to achieve this antiviral potency, in comparison with previous works where a large valency was required to reach similar activities.
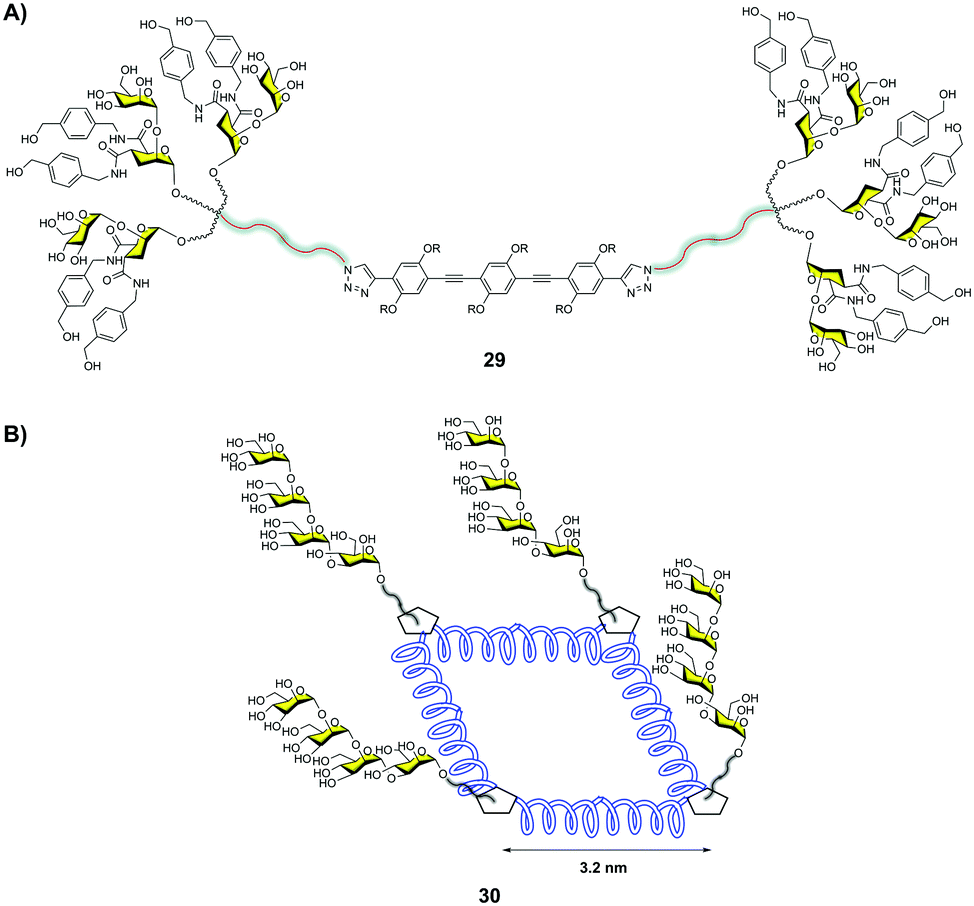 | ||
| Fig. 8 Hexavalent glycodendron with a rod central core 29 (A), and glycocluster with a square polyproline central core 30 (B). | ||
Very recently, Wang and co-workers39 designed a semirigid square scaffold based on tetra-helix polyproline. This scaffold presented four functionalization sites close to the corners, with estimated distances between them of around 3.2 nm (Fig. 8B). On these sites, trivalent glycodendrons decorated with different mannose oligosaccharides, such as a branched mannotriose or a linear Man4, were conjugated via the CuAAC reaction. These glycomacromolecules were evaluated by SPR to bind DC-SIGN and Langerin. In particular, the glycoconjugate with four trivalent glycodendrons of Man430 (Fig. 8B) showed a 4800-fold selectivity for DC-SIGN versus Langerin. This result could be explained based on the different distances between CRDs in the trimeric Langerin and the tetrameric DC-SIGN, 4.2 nm and 4 nm, respectively. This work highlighted how the control of glycan distances on a scaffold can provide strong selectivity to target DC-SIGN instead of Langerin with the relevant implications that this fact has for the design of antiviral compounds against HIV.
Most of the carbohydrate multivalent systems described to interact with DC-SIGN were based on simple mannosyl ligands up to tetrasaccharides. This was conditioned by the synthetic complexity of the natural ligand, the high mannose. Very few examples use Man9 as the ligand to prepare glycodendrimers to interact with DC-SIGN. Wong and co-workers described one of the first examples in 2008.40 A 2G dendron with nine copies of Man9 inhibited the binding of gp120 to DC-SIGN and the broadly neutralizing antibody 2G12 in the low micromolar to nanomolar range.
To overcome this problem, we developed a synthetic strategy to prepare the epitope Man9 of the high mannose.41 This straightforward synthesis provides the means to very efficiently obtain this branched nonasaccharide, paving the way for the preparation of multivalent systems. However, the bottleneck of the synthesis is the beta configuration of the mannose reducing end anomer, β-Man9. On the other hand, the natural beta configuration could have remarkable importance in the interaction with DC-SIGN when the Man9 epitope is in its rigid natural environment as Man9GlcNAc2-Asn, being part of a protein like the gp120 of HIV. However, this configuration when the Man9 epitope is coupled to scaffolds through flexible linkers could not have this relevance.
With this in mind and considering the much easier synthesis of the α-Man9, we prepared both anomers alpha 31 and beta 32 (Fig. 9) and compared their affinities for DC-SIGN.42 Using the corresponding α-Man9 and β-Man9 fluorescently labelled 33–34 (Fig. 9) in a fluorescence polarization assay, we demonstrated that both anomers interacted with DC-SIGN similarly. In other words, the configuration of the reducing end did not play a relevant role in the interaction when the epitope was not conjugated. In addition, we prepared the corresponding trivalent dendrons fluorescence-labelled 37–38 (Fig. 9). We observed that both trivalent dendrons had similar KD in the low μM range (0.53 and 0.37 for dendrons 37 and 38, respectively). This is one order of magnitude lower than the value observed for the corresponding monovalent nonasaccharides, showing a clear multivalent effect. Again, the anomeric configuration had no impact on the binding affinity of the trivalent systems. This fact supports the use of α-Man9 as a convenient ligand for DC-SIGN with the advantage of its preparation in comparison with its epimer β-Man9.
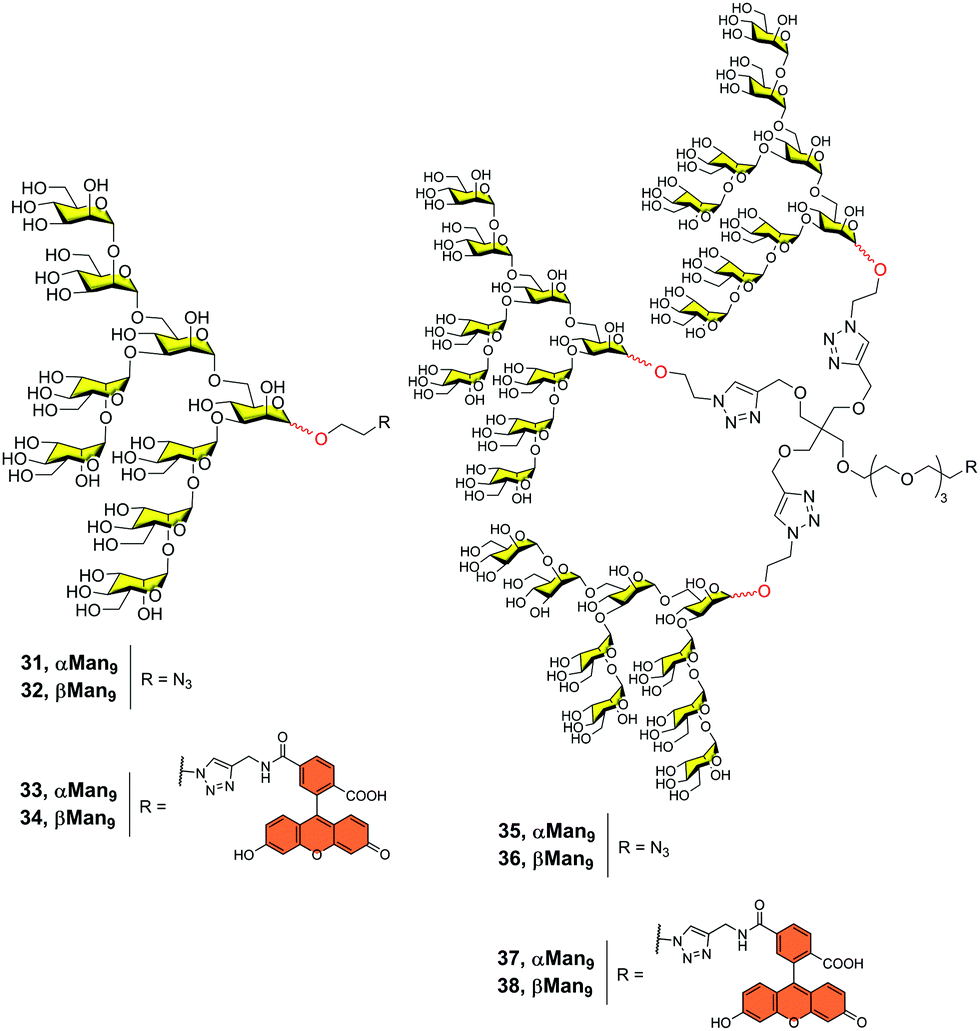 | ||
| Fig. 9 Structures of both α- and β-Man9 epitopes (31–34) and the corresponding trivalent dendrons unlabelled (35–36) and labelled (37–38) with a fluorescein tag. | ||
Mannosylated carbon nanoforms
In the search for innovative multivalent scaffolds, our research group has focused on bioactive carbon nanoforms developing over the last few years. Particularly, unexplored 3D fullerene C60 with highly symmetric geometry could be considered as an interesting biocompatible carbon platform for the multivalent presentation of carbohydrates. Mannosylated-functionalized fullerenes could mimic the carbohydrate cloak on the viral surface (generally of globular geometry), which can interfere with the infectious process by blocking the interaction with the DC-SIGN receptor. In this context, hexa-substituted glycofullerenes with Th octahedral symmetry and globular structure have been remarkably exploited during the last decade for the preparation of multivalent glycoconjugates.43,44Owing to the poor solubility of fullerene C60 in water, chemical functionalization is the starting point to integrate this nanomaterial into living systems (biocompatibility). One of the methodologies reported in the literature to obtain easily post-functionalizable hexakis-adducts consists of the Bingel–Hirsch addition of malonates to C60.45 Unlike other carbon-based platforms in which multivalent conjugation is much less defined, these post-functionalizable molecules allow the simultaneous grafting of twelve groups, which offers a clear advantage for the fast construction of dendritic structures.46 In 2010, our research group and the Martin group reported a straightforward strategy based on the CuAAC reaction47 to simultaneously “click” twelve carbohydrate residues, such as mannose, galactose, fucose, or a divalent glycodendron, to alkyne-substituted hexakis-adducts of fullerene C60 in a regioselective and efficient way in a few steps.48 In Fig. 10, the structure of the glycofullerene 39 coated with twelve mannoses is shown. Importantly, it was the first time fully water-soluble fullerene sugar balls were prepared in which the C60 core was completely grafted by sugar residues. It is worth noting that this facilitated the investigations of biological applications49–51 including enzymatic inhibitors,52–57 inhibition of bacteria,58–60 cancer,61,62 molecular recognition of Con A,63 as well as viral infections; the latter one will be discussed herein.
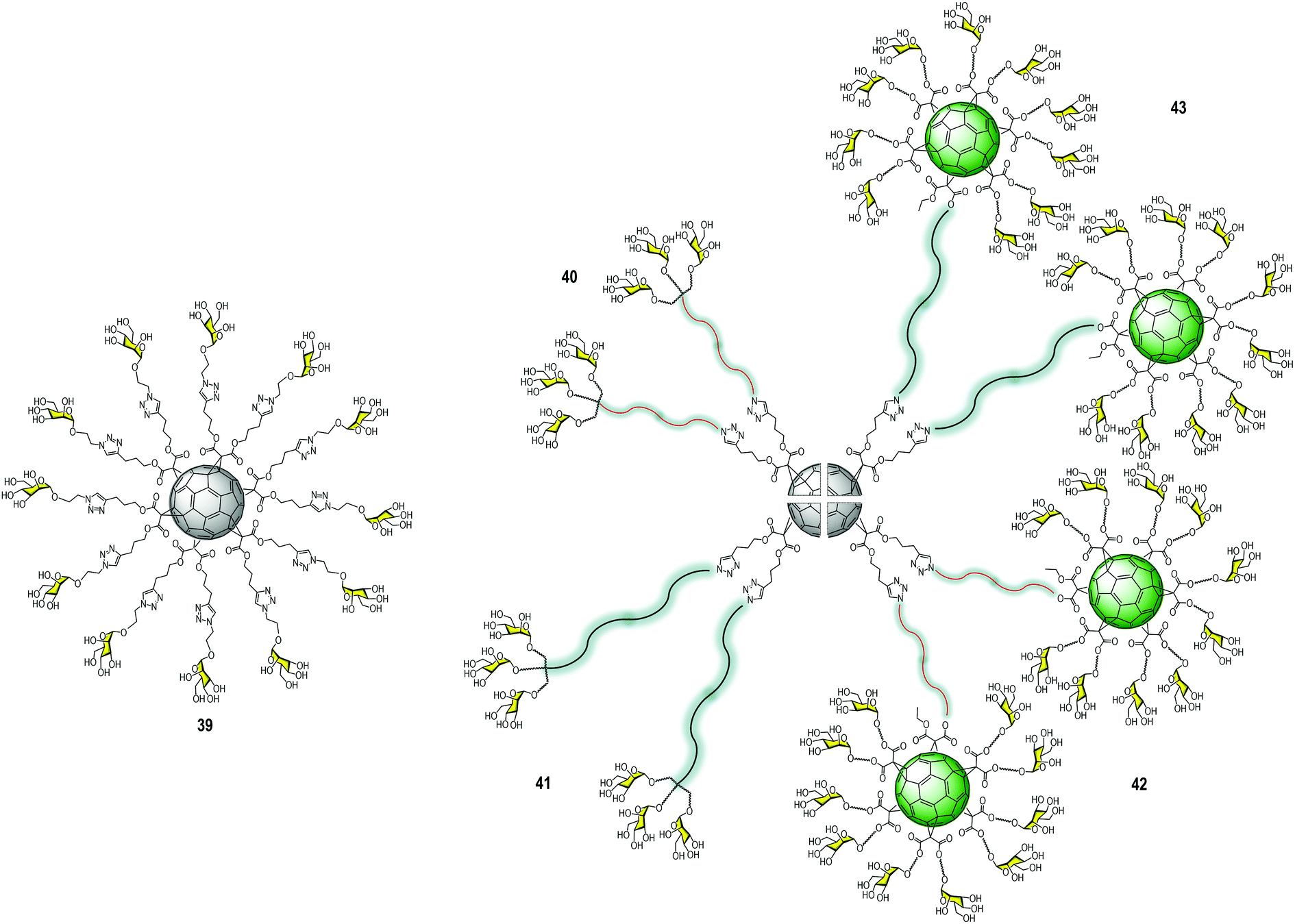 | ||
| Fig. 10 Structure of glycofullerene 39 (left) and general structures of glycodendrofullerenes 40–41 and tridecafullerenes 42–43 (right). | ||
Using the same convergent strategy, for the first time, we have prepared glycodendrofullerenes as antiviral agents against the Ebola virus infection, in which the fullerene C60 core was surrounded by small mannosylated glycodendrons.64 In this study, we carried out the synthesis of two glycodendrofullerenes 40–41 (Fig. 10) bearing 36 copies of mannoses in the periphery, employing CuBr·S(CH3)2 as a catalyst in presence of a piece of metallic Cu, ensuring the presence of Cu0 in the reaction media. Although these two glycodendrofullerenes contain the same number of carbohydrates, two different spacers between the central C60 core and the peripheral carbohydrate-substituted dendron were used. This allowed us to study the effects of the steric congestion of these glycofullerenes on their antiviral properties. We postulated that the most flexible and longer ethylene glycol-based linker (compound 41) could allow better accessibility and availability of the carbohydrates in the interaction with DC-SIGN. A simple FTIR analysis indicated the absence of the typical bands for alkyne and azide groups (at ∼2117 and 2092 cm−1, respectively) in the starting materials, indicating the efficiency of the cycloaddition step. Most importantly, the 13C NMR characterization of this glycofullerene derivative with octahedral symmetry was particularly useful for demonstrating the full functionalization of the alkyne residues. The absence of the typical signals corresponding to the alkyne groups (at ∼69.0 and 83.0 ppm) in the fullerene precursor and the signal of the CH2 bound to the azido group at ∼50 ppm in the glycodendron precursors in the NMR spectra corroborated that both the cycloaddition step and the subsequent purification were efficiently carried out. On the other hand, only two signals appeared for the sp2 carbons (at ∼141 and 145 ppm) and a signal at δ ∼ 69 for the two sp3 carbons of the C60 core, providing evidence of the high Th symmetry of the compound.
Based on the success of this synthetic strategy and in order to dramatically increase the valency and the size of the previous glycofullerenes, we have reported the synthesis of tridecafullerenes, the so-called “superballs”.65 These superballs bearing 120 peripheral sugar ligands are constituted by a central C60 core in which the 12 alkyne moieties were clicked to 10 sugar-containing [60]fullerene units. In the same way as glycodendrofullerenes, two mannosylated tridecafullerenes (42–43) with different spacers between the peripheral sugar-coated fullerenes and the C60 central core were prepared. (Fig. 10) Also, a tridecafullerene was coated with 120 copies of galactose as a negative control for the biological assays since DC-SIGN is not able to recognize this sugar.
Despite the enormous size and molecular complexity, the synthesis of superballs was straightforward (in four synthetic steps) and, most importantly, easily reproducible using our convergent strategy. The synthesis relied on the grafting of a clickable asymmetric [5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1]-hexaadduct (A10B macromonomer) onto the hexakisadduct of fullerene C60 core 50. As a typical example, the preparation of tridecafullerene 42 is depicted in Fig. 11. The synthesis started with the preparation of monoadduct 44, which resulted from the Bingel–Hirsch nucleophilic monoaddition of 6-bromohexyl ethyl malonate to fullerene C60.66 Subsequent Bingel–Hirsch nucleophilic cyclopropanation of 44 and di(pent-4-yn-1-yl) malonate (45) in the presence of CBr4 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as base gave the A10B macromonomer 46 decorated with 10 alkynes and a bromine atom at the focal point. After that, 46 was subjected to the CuAAC reaction with 2-azidoethyl α-D-mannopyranoside (47) using CuBr·S(CH3)2 as the catalyst and sodium ascorbate as the reducing agent in the presence of a piece of metallic Cu, to give derivative 48 in good yield. Replacement of the bromine by an azide group, followed by click reaction to the symmetric hexa-adduct 50 under the same CuAAC conditions, afforded the giant superball 42 with ∼70% yield, after the removal of copper and purification by precipitation in the reaction medium. It is important to highlight that these water-soluble giant globular multivalent glycofullerenes represent the first example of the fastest dendrimeric growth reported at that time.
:1]-hexaadduct (A10B macromonomer) onto the hexakisadduct of fullerene C60 core 50. As a typical example, the preparation of tridecafullerene 42 is depicted in Fig. 11. The synthesis started with the preparation of monoadduct 44, which resulted from the Bingel–Hirsch nucleophilic monoaddition of 6-bromohexyl ethyl malonate to fullerene C60.66 Subsequent Bingel–Hirsch nucleophilic cyclopropanation of 44 and di(pent-4-yn-1-yl) malonate (45) in the presence of CBr4 and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as base gave the A10B macromonomer 46 decorated with 10 alkynes and a bromine atom at the focal point. After that, 46 was subjected to the CuAAC reaction with 2-azidoethyl α-D-mannopyranoside (47) using CuBr·S(CH3)2 as the catalyst and sodium ascorbate as the reducing agent in the presence of a piece of metallic Cu, to give derivative 48 in good yield. Replacement of the bromine by an azide group, followed by click reaction to the symmetric hexa-adduct 50 under the same CuAAC conditions, afforded the giant superball 42 with ∼70% yield, after the removal of copper and purification by precipitation in the reaction medium. It is important to highlight that these water-soluble giant globular multivalent glycofullerenes represent the first example of the fastest dendrimeric growth reported at that time.
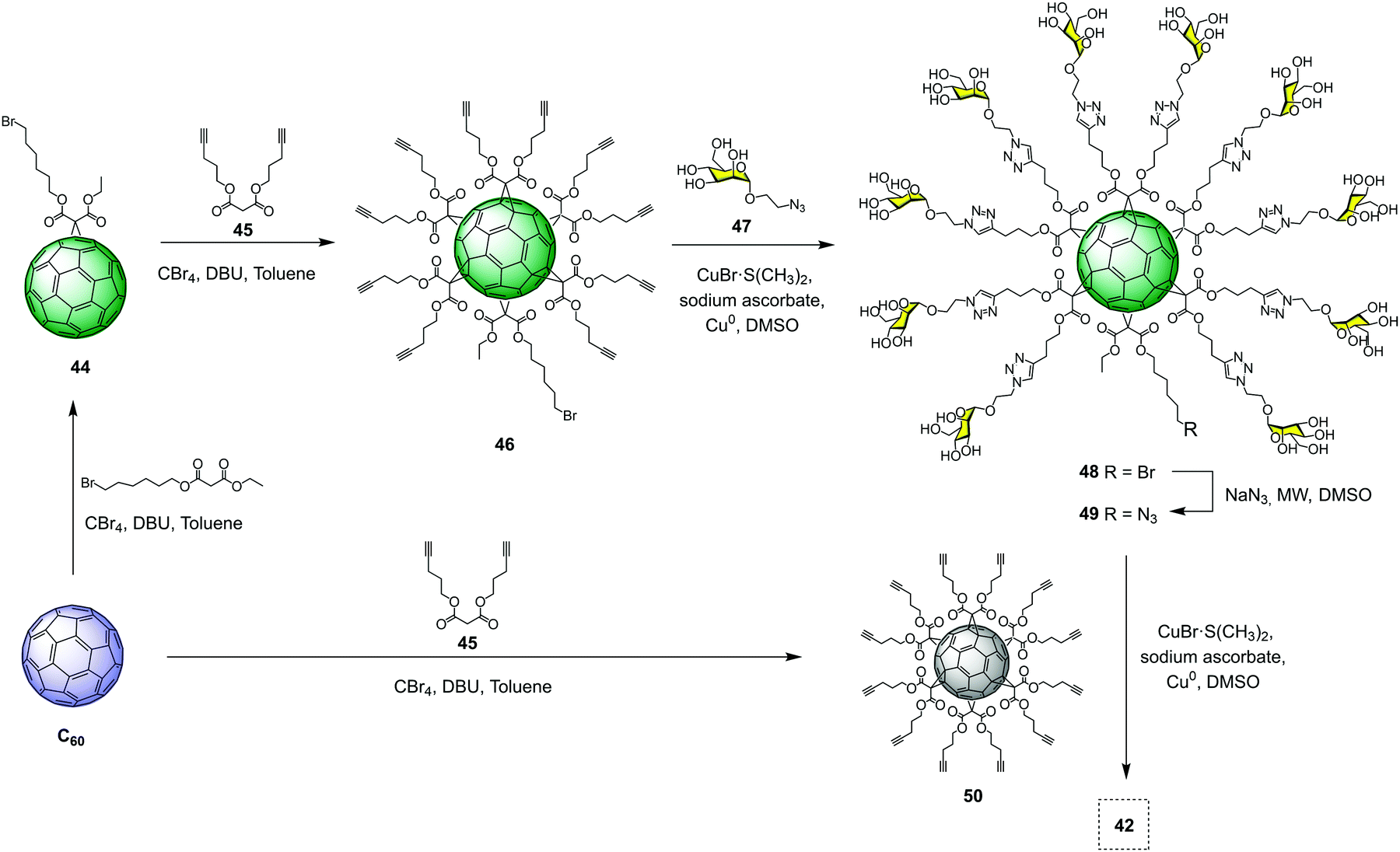 | ||
| Fig. 11 Synthesis of tridecafullerene 42. | ||
Notwithstanding the molecular weights as high as 56 kDa, the tridecafullerenes 42–43 were fully characterized by FTIR and NMR spectroscopy as well as dynamic light scattering (DLS), transmission electron microscopy (TEM) and X-ray photoelectron spectroscopy (XPS). Regarding FTIR and 13C NMR spectroscopy, we observed the same typical signals as previously mentioned for glycodendrofullerenes. The morphology of these superballs was determined by TEM experiments as small spherical particles with a size of ∼4 nm, which was in good agreement with the experimental findings in DLS analyses. The nature, as well as relative abundance of the atoms present, were measured by XPS analysis. In all cases, the expected elements (C 1s, O 1s, and N 1s) according to their relative abundance were shown. These data corroborated that both the cycloaddition step and the subsequent purification were efficiently carried out.
The ability of these glycodendrofullerenes and tridecafullerenes to inhibit the infection of Jurkat-DC-SIGN cells using Ebola virus glycoprotein (EBOGP) pseudotyped viral particles as infectious agents was evaluated (Table 1). In addition to those, glycofullerene decorated with 12 mannoses 39,63 as well as α-methyl-D-mannopyranoside were also included in these series to study the effect of multivalency presentation on the antiviral properties.
| Compound | No. of Man | IC50 (nM) | RIPa |
|---|---|---|---|
| a RIP, calculated as (IC50)mono/IC50 × valency (IC50)mono, IC50 of the monovalent compound; IC50 × valency, IC50 of the multivalent compound multiplied by the number of ligands present in the glycofullerene compound. | |||
| 43 | 120 | 0.667 | 1.58 × 104 |
| 42 | 120 | 20.375 | 5.2 × 102 |
| 41 | 36 | 300 | 1.17 × 102 |
| 40 | 36 | 68000 |
0.5 |
| 39 | 12 | 2000 | 53 |
| α-Methyl-D-mannopyranoside | 1 | 1.27 × 106 | 1 |
All glyconanostructures showed no cytotoxicity in cell lines at the concentrations used in the infection experiments, allowing the study of their potential biological function in preventing viral infection. As expected, the results obtained in these experiments revealed the dependence of the inhibition effect on the mannoses. The galactosylated version of the tridecafullerene was not able to inhibit the infection process mediated by DC-SIGN. A multivalent effect was observed when glycofullerene 39 was compared with glycodendrifullerene 41 (more than 6-fold increase), whereas the most compact 40 showed 34-fold less activity than the fullerene with 12 mannoses. Regarding both tridecafullerenes 42 and 43, these studies revealed that they could effectively block EBOV infection at the nanomolar to picomolar concentration range. The exhibited strong antiviral activity for tridecafullerene 43 (long linker) was more than one order of magnitude more potent in comparison with superball 42 (short linker) with an IC50 of ∼0.7 and ∼20 nM, respectively. The IC50 values for tridecafullerenes were surpassed by four and three orders of magnitude in comparison with glycofullerene 39 and glycodendrofullerene 41 endowed with 12 and 36 mannoses, respectively. These data revealed that the length and the flexibility of the linker unit and consequently the adequate accessibility of the ligands to interact with DC-SIGN, as well as valency number, could be considered important factors to take into account for obtaining high affinity towards antiviral activities. The importance of the length of the linker in glycofullerenes in biomedical applications was previously confirmed by NMR spectroscopy studies.67,68
These results reported by our group produced a huge impact in the scientific community, encouraging other research groups to employ these glycodendrofullerenes and tridecafullerenes coated with other sugars for biological applications. In this context, Nierengarten et al.69,70 reported the synthesis of giant glycofullerenes coated with iminosugars as potential glycosidase inhibitors.
To check if other glycofullerenes with different sizes and geometries showed an improvement in the biological antiviral activity, we explored glycodendrofullerenes with amphiphilic character using C60 mono-adducts instead of C60 hexakis-adducts.71 These amphiphilic dendrofullerenes in aqueous media allowed for self-assembly, obtaining supramolecular arrangements ranging from nanorods to nanovesicles, as we reported previously.72 Monoadducts of fullerene C60 functionalized with two and four trivalent mannose glycodendrons were obtained following the optimized CuAAC methodology previously reported. Scanning electron microscopy (SEM) images in water and XRD and low-angle X-ray scattering (SAXS) experiments in powder revealed that spherical micelle-type aggregates with a similar and uniform size were observed for both glycodendrofullerenes (Fig. 12). The data suggested strong π–π interactions between fullerene C60 units leading to these supramolecular aggregates. Both micellar structures of globular symmetry showed the ability to inhibit DC-SIGN in the range of nanomolar concentrations in the Ebola virus experimental model.71
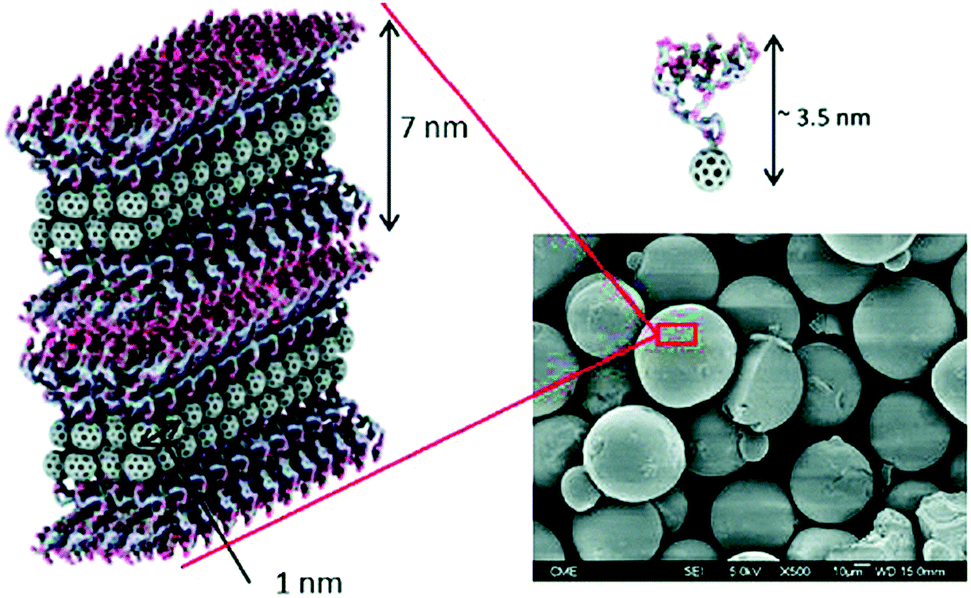 | ||
| Fig. 12 SEM micrograph of glycodendrofullerene with 6 mannoses, where the compact internal structure of the micelles is shown. Image reproduce with permission from the Royal Society of Chemistry.71 | ||
In addition to fullerenes C60, a large variety of carbon nanoforms, such as single and multi-walled carbon nanotubes (SWCNTs/MWCNTs), single-walled carbon nanohorns (SWCNHs) and more recently 2D graphene, graphene quantum dots (GQD) and carbon quantum dots (CQD)73 have also been developed for applications in biological fields.74–81 Different methodologies for their surface functionalization have been developed,82–85 providing new properties, and preventing toxicity by the asbestos-like behavior they typically display.86,87 Particularly, we focused our attention on SWCNTs, MWCNTs and SWCNHs as scaffolds to generate multivalent nanosized glycoconjugates as potential antiviral agents.88 These carbon nanoforms were chosen as platforms for sugar presentation because they could mimic the shape or the surface of the Ebola virus, since the elongated shape of CNTs resembles the filamentous structure of this virus, characteristic of a thread virus. Therefore, we could study the influence of the shape and size of these platforms to interact with DC-SIGN in comparison to the fullerene C60 derivatives synthesized previously.
As a proof of concept, we first validated the use of mannosylated SWCNTs for selective carbohydrate–lectin interactions.89 For this purpose, we have reported the synthesis of glycoSWCNTs by the covalent functionalization of SWCNTs and trivalent α-D-mannosyl dendrons 24a. These hybrid systems demonstrated selective interactions with ConA, as proved by fluorescence, AFM and UV-vis studies.
In 2018, we reported the preparation of glyconanomaterials constituted by glycofullerene 49 or glycodendron 4 containing ten and nine copies of mannoses, respectively, covalently attached to SWCNT, MWCNT and SWCNH carbon nanoforms as multivalent scaffolds to obtain a set of glycoconjugates 51–56 (Fig. 13).88 Apart from size and shape, these allowed us to study whether the presence of both glycofullerenes and glycodendrons could have an effect on the biological activity of these hybrid glyconanomaterials.
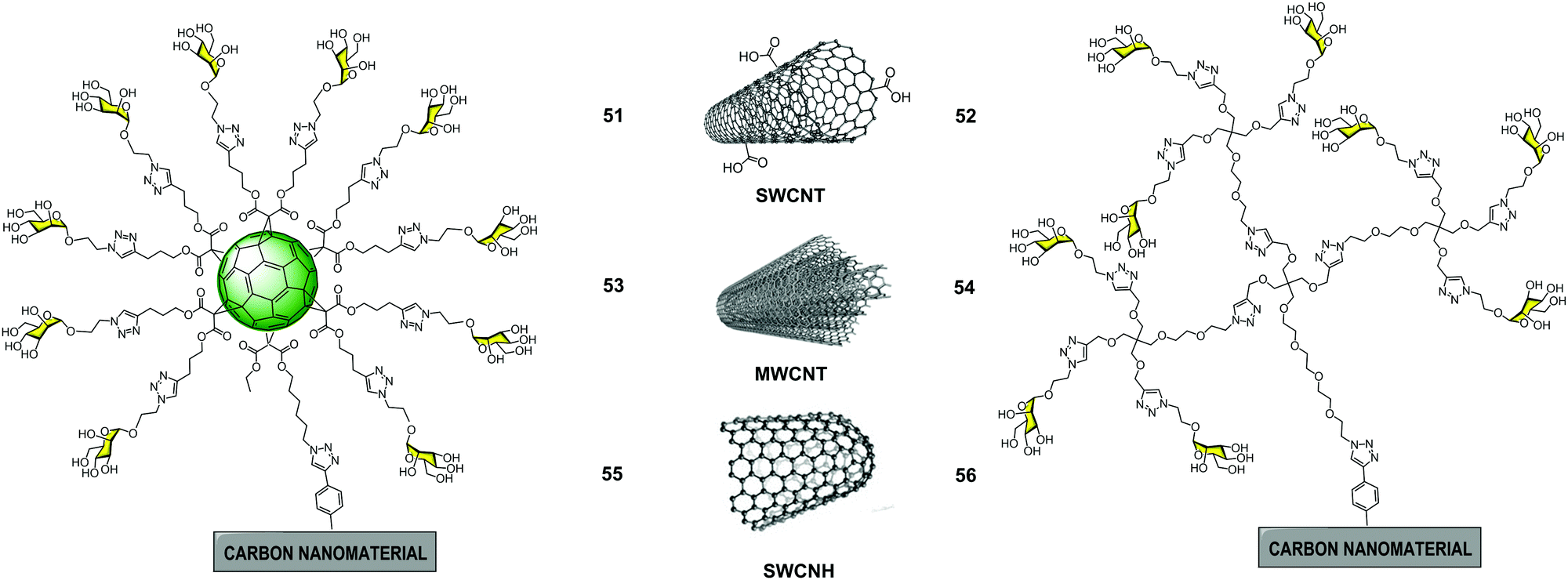 | ||
| Fig. 13 General structures of glyconanomaterials 51–56 functionalized with glycofulleres (left) and glycodendrons (right). | ||
The synthetic procedure for obtaining the glyconanomaterials 51–56 was carried out in an analogous way for all the carbon nanoforms. In a first step, carbon nanostructures were functionalized with protected phenylethynyl groups by a Tour reaction90,91 to install the required alkyne for the CuAAC reaction. Later, in a second step, one pot deprotection of the terminal alkynes and a CuAAC click reaction with the corresponding azide-substituted glycofullerene 49 and glycodendron 4 were accomplished. The CuAAC reaction was carried out following the optimised methodology previously reported. A galactose derivative formed by SWCNH and galactosyl glycofullerene, was synthesized in an analogous manner and used as a negative control in the biological assays. The characterization of these hybrid materials, as well as the key intermediates, using conventional techniques applied in material sciences such as thermogravimetric analysis (TGA), Raman spectroscopy, FTIR, DLS, XPS and TEM, provided insights into the physicochemical properties of the materials.
The antiviral activities of glycanomaterials 51–56 decorated with mannose residues were evaluated using the same system of pseudotyped viral particles that present EBOV glycoproteins on their surface in a similar manner as described previously. The experimental finding revealed that for each type of carbon nanoform, the most active glycoconjugate was that with a higher mannose content. On the other hand, it was found that both the size and morphology of the scaffold used for the multivalent presentation of ligands were important. A clear multivalent effect was observed for glycoconjugates based on SWCNTs and MWCNTs when compared to glycofullerene 39 and glycodendron 4; the latter did not show any antiviral activity at the concentrations investigated. In particular, glyconanomaterials based on 3D MWCNTs 53–54 were the best inhibitors of EBOV infection (IC50 of 0.37 μg mL−1 for 53 and 1.90 μg mL−1 for 54), with no appreciable cytotoxic effects. Based on these results, carbon nanoforms can be considered as efficient multivalent carbon-based platforms with tunable size and high biostability properties that make them very appealing materials for biomedical applications.
More recently, our research group developed new superstructures as potential inhibitors in the infection of emergent viruses such as Zika (ZIKV) and Dengue (DENV) viruses. In this context, we reported the preparation of groundbreaking α(1,2)mannobiosylated tridecafullerenes, so-called “nanoballs”, decorated with 12 to 360 carbohydrates units (Fig. 14).92 The α(1,2)mannobioside was chosen instead of mannose, since the affinity of the former for DC-SIGN increased by 3–4 fold in comparison to the corresponding monosaccharide.10,93,94 Regarding the synthesis of the α(1,2)mannobioside conveniently functionalized with an azido group in the anomeric position, this was performed using the methodology reported by us to obtain this disaccharide in an efficient manner.41,95 Unlike glycofullerenes reported previously, we decided to employ a synthetic strategy based on metal-free click chemistry, namely, the strain-promoted cycloaddition of azides to alkynes (SPAAC). From a synthetic point of view, the SPAAC reaction presents clear advantages in comparison with the CuAAC version, such as easier purification, lower reaction times, higher yields and, most importantly, the absence of high cytotoxic copper, which is a serious drawback. The use of copper can also affect the click reaction, lowering the yields and even, in some cases, inhibiting the reaction96 due to the chelating ability of the triazole rings and, in this particular case, of the α(1,2)mannobiosides, since chelating copper between the two monosaccharide moieties could occur.
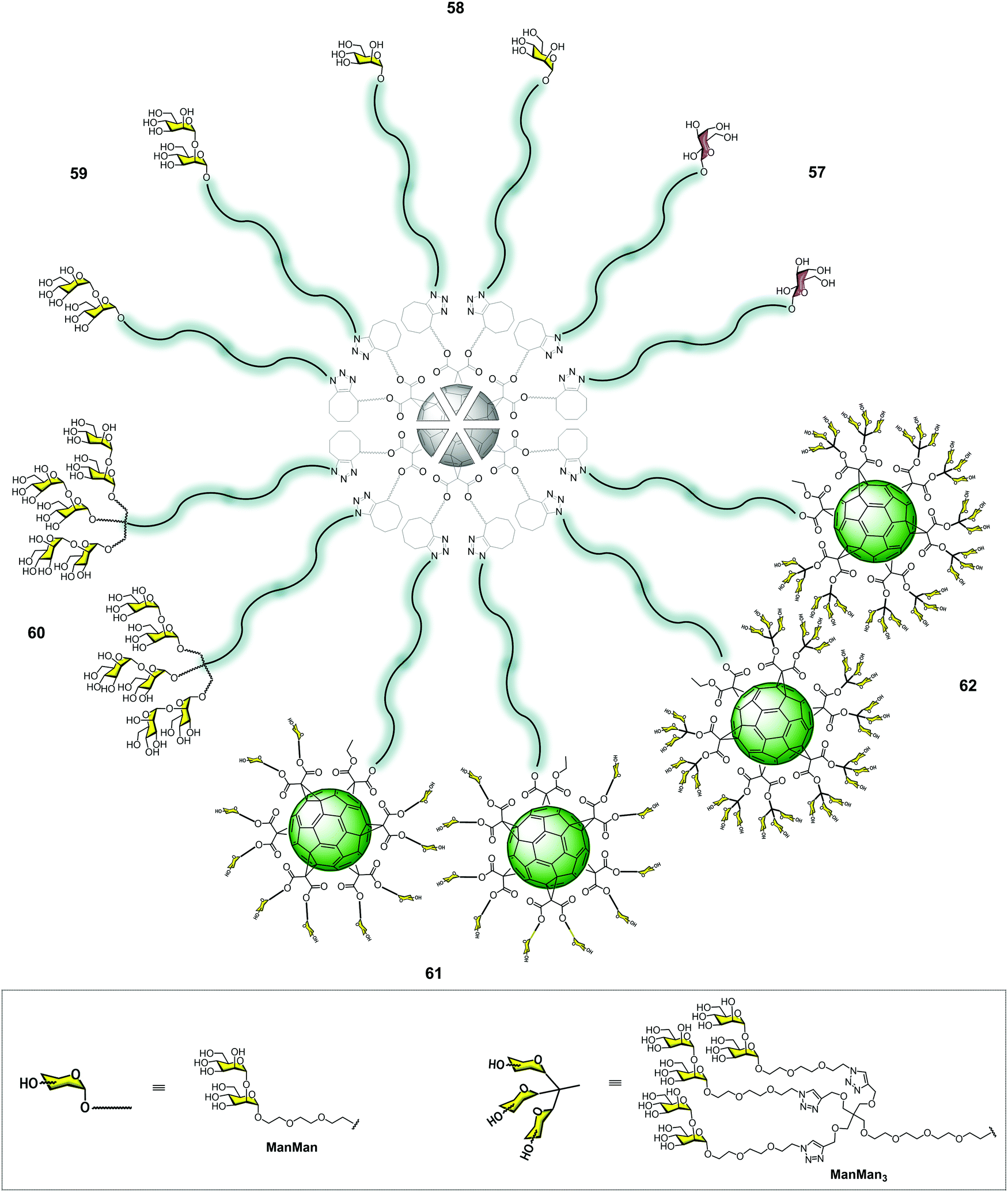 | ||
| Fig. 14 Cartoon representing the general structures of glycodendrons appended with 12 galactose (57), mannose (58) and α(1,2)mannobiosides (59), glycodendrofullerene with 36 α(1,2)mannobiosides (60) and nanoballs with 120 (61) and 360 (62) α(1,2)mannobiosides. | ||
Firstly, we carried out the synthesis of post-functionalizable symmetric and asymmetric hexakis-adducts of fullerene C60 with twelve and ten cyclooctyne groups, respectively.97,98 In order to confirm the versatility of the highly symmetric hexakis adducts of [60]fullerene, conjugation via the SPAAC reaction with different azides, including a natural product, an amino acid or a peptide nucleic acid (PNAs) monomer, was satisfactorily tested.97
In contrast to the CuAAC strategy, the SPAAC methodology allowed for the efficient assembly of glycofullerenes, glycodendrofullerenes and nanoballs by heating the reaction mixture at 50 °C under microwave irradiation for 30 min in excellent yields. In this way, we obtained glycofullerenes functionalized with 12 galactoses (57, as the negative control), 12 mannoses (58) and 12 α(1,2)mannobiosides (59) and glycodendrofullerene (60) with 36 α(1,2)mannobiosides motifs (Fig. 14). In order to dramatically increase the multivalency of these mannobiosylated glycostructures, we decided to carry out the preparation of groundbreaking nanoballs coated with 120 (61) and 360 (62) α(1,2)mannobiosides residues (Fig. 14). It is worth highlighting that the synthesis of the derivative containing 360 disaccharide units, obtained in a synthetic step, represents the fastest dendritic growth reported in the literature, which has 41.370 atoms (C, H, O, N). Similar to the previously mentioned superballs, the synthesis of the nanoballs 61–62 relies on the grafting of a clickable A10B macromonomer onto the symmetric hexakisadduct of the previously used fullerene C60 core.
Regarding the inhibitory effect of these molecules, they were evaluated in a pseudotyped Zika and Dengue viral infection model employing Jurkat DC-SIGN cells. In these experiments, the dependence on the number of sugars to inhibit DC-SIGN was observed, as expected. The antiviral activity of the three most promising compounds with 36 (60), 120 (61) and 360 (62) disaccharides was measured (Table 2).
| Compound | No. of Man–Man | IC50 (nM) Zika | IC50 (nM) Dengue |
|---|---|---|---|
| 60 | 36 | 8.35 | 7.71 |
| 61 | 120 | 0.52 | 0.098 |
| 62 | 360 | 0.067 | 0.035 |
The experimental data revealed that the three nanoballs 60–62 showed very strong antiviral activity at nanomolar to picomolar concentrations for both Zika and Dengue viruses, while the glycofullerene decorated with galactoses did not exhibit such activity. In particular, nanoball 62 bearing 360 disaccharides was the best at inhibiting the viral infection for both viruses, with an IC50 of 67 pM for Zika and 35 pM for Dengue. Moreover, a clear multivalent effect was observed with the increase of the carbohydrate units, showing an enhancement of one order of magnitude. The data suggested that an increase in the valency of the systems achieved improved antiviral activity, validating the use of nanoballs as antiviral agents as effective probes to block carbohydrate receptors and inhibit the infection process. To the best of our knowledge, the nanoball surrounded by 360 carbohydrate units is the most efficient inhibitor of ZIKV and DENV infections reported so far, with an IC50 in the picomolar range.
Other mannosylated scaffolds
Nanoparticles, in particular gold nanoparticles, have been commonly used as a platform for the multivalent presentation of carbohydrates.20,99 The control of the size, the facility to attach sugars using terminal thiols, the globular presentation of the sugars on a large surface, and the high multivalency that this platform offers has made this approach very popular for developing glycotools to interact with lectins.100 The nanoparticle surface decorated with carbohydrates mimics the presentation of glycosphingolipids at the cell surface as a dense coat covering a large area. However, few examples combine glycodendrons with gold nanoparticles.101We reported the construction of gold nanoparticles (GNPs) with zwitterionic ligands combined with mannosyl trivalent glycodendrons to create glycodendronanoparticles 63 (Fig. 15).102 For this purpose, a classical method for generating gold nanoparticles (GNPs) was used.103 The in situ reduction of a gold salt (HAuCl4) with NaBH3 in the presence of the thiol ligands provided gold cores with diameters smaller than 2 nm. This small size of the metal core produces an intense red to near-infrared fluorescence.
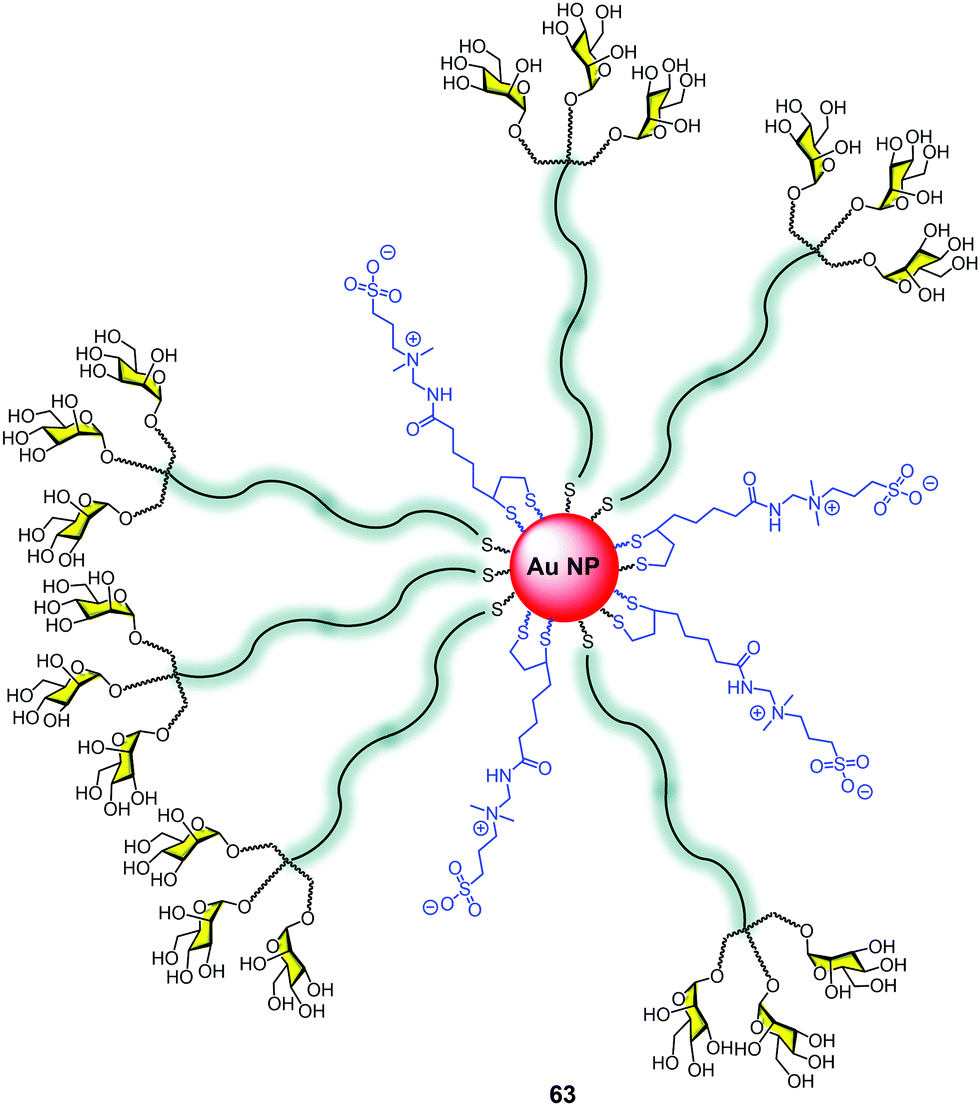 | ||
| Fig. 15 GNP with mannosyl trivalent dendrons and the zwitterionic ligand (63). | ||
The uptake of these GNPs by human dendritic cells (hDCs) was 2.5-fold better than the corresponding to GNPs without sugars. Moreover, the use of mannan as a C-type lectin (MR, DC-SIGN) inhibitor decreased the uptake to 60%, demonstrating the relevance of the sugars in facilitating the uptake by hDCs via a specific receptor.
The idea behind the synthetic mannosylated multivalent dendrimers is the creation of mimetics as close as possible to the natural target. Considering the presentation of sugars at the surface of pathogens such as HIV, we evaluated the possibility of using the capsid of a virus as an ideal platform to mimic this carbohydrate presentation on pathogen surfaces. In collaboration with the Davis group, we reported the preparation of well-defined virus-like glycodendriparticles.68 For this goal, we chose the bacteriophage Qβ-coated protein that spontaneously self-assembled to incorporate 180 units in a big sphere with a diameter of around 28 nm. Using site-directed mutagenesis, L-homopropargylglycines (Hpg) were incorporated in place of the wild-type methionine. This non-natural amino acid provided an alkyne group as a functional side chain for coupling. Trivalent 24a and nonavalent 4 mannosylated dendrons were conjugated to this scaffold via a CuAAC reaction to yield monodisperse glycodendriproteins with 540 (64a) and 1620 (64b) mannoses, respectively (Fig. 16). They presented an average distance between glycan residues of around 2.8–4.0 nm.
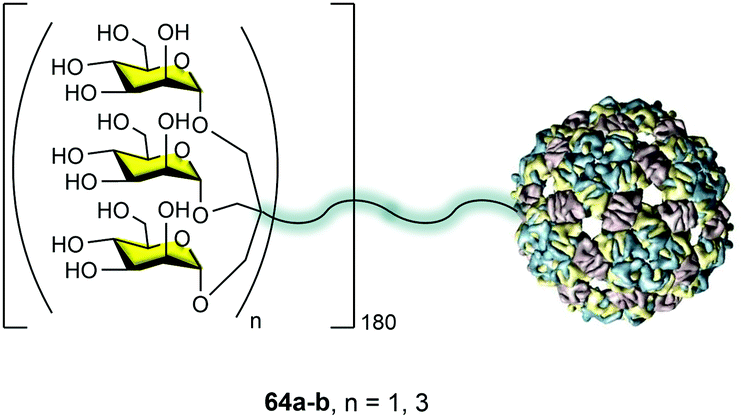 | ||
| Fig. 16 Glycodendriproteins with 540 (64a, n = 1) and 1620 (64b, n = 3) mannoses. | ||
It is important to highlight that the coupling of the glycodendrons (180 clicks simultaneously) was extremely efficient and all the alkynes reacted to incorporate the glycodendrons. This fact was demonstrated by the treatment of the protein with the reducing agent tris(2-carboxyethyl)phosphine (TCEP), which reduced the disulfide bonds, recovering the 180 monomers. These monomers were analysed by LC-ESI-MS showing that all were conjugated with a glycodendron, confirming the whole functionalization of the system.
These glycodendriproteins showed strong inhibitory activity in a pseudotyped Ebola virus infection model using Jurkat-DC-SIGN as well as monocyte-derived dendritic cells. These systems bearing 180 glycodendrons of mannose inhibited the infection process, with IC50 in the nanomolar to picomolar range. This example highlighted the power of this strategy that uses a viral capsid modified at will with glycodendrons to create a particle capable of being a pathogen competitor, inhibiting cell infection.
Outlook
Since the discovery of DC-SIGN, many contributions have provided a lot of information and progress in this field. Considering that DC-SIGN is a new target that appeared only two decades ago, remarkable improvements have been achieved concerning its expression and accessibility, understanding the selectivity that it shows toward carbohydrates, its biological role, and the design and development of agonist and antagonist molecules. These studies have provided an ideal scenario for developing the synthetic capacity of chemists creating amazing structures based on convergent strategies such as those described here. The approaches to synthesizing these carbohydrate multivalent systems are diverse and take advantage of the synthetic arsenal available to address the challenges of preparing these complex molecules.These systems are very valuable tools for exploring the binding of this lectin, to analyse the internalization processes, as well as the intracellular signalling, shedding light on those relevant processes related to the immune system where this receptor is involved. Based on the aforementioned results, multivalency, the nature of ligands and the shape and size of the platform, as well as the presentation and accessibility of carbohydrates, all seem to be very important factors for interaction with DC-SIGN. Small systems with a low valency but an adequate design to reach at least two CRDs on the same DC-SIGN tetramer have been demonstrated as being very efficient for achieving inhibitory capacities in the nanomolar range.
Some of these structures have shown a strong capacity to block DC-SIGN, inhibiting the infection process of different viruses such as HIV-1, Ebola, Dengue, or Zika. This ability to block DC-SIGN opens the door for their application as anti-pathogen agents. Despite all the achievements during these two decades with carbohydrate dendritic compounds showing a remarkable inhibitory activity in vitro, to our best knowledge, nowadays, there are no ongoing clinical or preclinical studies. The reason for this could be found in the complexity of these compounds, the cost of their preparation on a large scale for these studies, and also because they are far away from the classical concept of a drug. This last argument may not be a compelling reason due to the increasing popularity of biologicals as drugs, which have achieved great success for the treatment of rheumatoid arthritis, among other diseases. We consider that the chance to have a candidate enter clinical studies could be based on more accessible carbohydrate multivalent systems with a low valency but an appropriate design. However, many issues such as the cytotoxicity, druggability, bioavailability, and effectivity in animal models, must be addressed before these systems can be considered as candidates to enter clinical trials.
Despite all the knowledge accumulated in this field, many interesting contributions are still expected to come in the following years to complete the information related to this lectin and its role in triggering an immune response.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by MINECO (CTQ2017-86265-P) and Junta de Andalucía (Postdoctoral contract of JRS).Notes and references
- B. M. Curtis, S. Scharnowske and A. J. Watson, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 8356–8360 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek, D. S. Kwon, R. Torensma, S. J. van Vliet, G. C. F. van Duijnhoven, J. Middel, I. L. M. H. A. Cornelissen, H. S. L. M. Nottet, V. N. KewalRamani, D. R. Littman, C. G. Figdor and Y. van Kooyk, Cell, 2000, 100, 587–597 CrossRef CAS.
- T. B. H. Geijtenbeek, R. Torensma, S. J. van Vliet, G. C. F. van Duijnhoven, G. J. Adema, Y. van Kooyk and C. G. Figdor, Cell, 2000, 100, 575–585 CrossRef CAS.
- Y. van Kooyk and T. B. H. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697–709 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek and Y. van Kooyk, APMIS, 2003, 111, 698–714 CrossRef CAS PubMed.
- Y. van Kooyk, A. Engering, A. N. Lekkerkerker, I. S. Ludwig and T. B. Geijtenbeek, Curr. Opin. Immunol., 2004, 16, 488–493 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek, J. den Dunnen and S. I. Gringhuis, Future Microbiol., 2009, 4, 879–890 CrossRef CAS PubMed.
- F. Zhang, S. Y. Ren and Y. F. Zuo, Int. Rev. Immunol., 2014, 33, 54–66 CrossRef CAS PubMed.
- D. A. Mitchell, A. J. Fadden and K. Drickamer, J. Biol. Chem., 2001, 276, 28939–28945 CrossRef CAS PubMed.
- H. Feinberg, D. A. Mitchell, K. Drickamer and W. I. Weis, Science, 2001, 294, 2163–2166 CrossRef CAS PubMed.
- K. J. Doores, C. Bonomelli, D. J. Harvey, S. Vasiljevic, R. A. Dwek, D. R. Burton, M. Crispin and C. N. Scanlan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13800–13805 CrossRef CAS PubMed.
- H. Feldmann, S. T. Nichol, H. D. Klenk, C. J. Peters and A. Sanchez, Virology, 1994, 199, 469–473 CrossRef CAS PubMed.
- E. van Liempt, C. M. C. Bank, P. Mehta, J. J. Garcia-Vallejo, Z. S. Kawar, R. Geyer, R. A. Alvarez, R. D. Cummings, Y. van Kooyk and I. van Die, FEBS Lett., 2006, 580, 6123–6131 CrossRef CAS PubMed.
- M. J. Borrok and L. L. Kiessling, J. Am. Chem. Soc., 2007, 129, 12780–12785 CrossRef CAS PubMed.
- A. Cambi, F. de Lange, N. M. van Maarseveen, M. Nijhuis, B. Joosten, E. M. H. P. van Dijk, B. I. de Bakker, J. A. M. Fransen, P. H. M. Bovee-Geurts, F. N. van Leeuwen, N. F. Van Hulst and C. G. Figdor, J. Cell Biol., 2004, 164, 145–155 CrossRef CAS PubMed.
- G. Tabarani, M. Thepaut, D. Stroebel, C. Ebel, C. Vives, P. Vachette, D. Durand and F. Fieschi, J. Biol. Chem., 2009, 284, 21229–21240 CrossRef CAS PubMed.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- M. Sanchez-Navarro and J. Rojo, Drug News Perspect., 2010, 23, 557–572 CrossRef CAS PubMed.
- J. J. Reina and J. Rojo, Braz. J. Pharm. Sci., 2013, 49, 109–124 CrossRef CAS.
- M. Marradi, F. Chiodo, I. Garcia and S. Penades, Chem. Soc. Rev., 2013, 42, 4728–4745 RSC.
- N. Jayaraman, K. Maiti and K. Naresh, Chem. Soc. Rev., 2013, 42, 4640–4656 RSC.
- I. Pramudya and H. Y. Chung, Biomater. Sci., 2019, 7, 4848–4872 RSC.
- K. Vong, T. Yamamoto and K. Tanaka, Small, 2020, 16 Search PubMed.
- E. Arce, P. M. Nieto, V. Diaz, R. G. Castro, A. Bernad and J. Rojo, Bioconjugate Chem., 2003, 14, 817–823 CrossRef CAS PubMed.
- F. Lasala, E. Arce, J. R. Otero, J. Rojo and R. Delgado, Antimicrob. Agents Chemother., 2003, 47, 3970–3972 CrossRef CAS PubMed.
- G. Tabarani, J. J. Reina, C. Ebel, C. Vives, H. Lortat-Jacob, J. Rojo and F. Fieschi, FEBS Lett., 2006, 580, 2402–2408 CrossRef CAS PubMed.
- J. Rojo and R. Delgado, J. Antimicrob. Chemother., 2004, 54, 579–581 CrossRef CAS PubMed.
- R. Ribeiro-Viana, J. J. Garcia-Vallejo, D. Collado, E. Perez-Inestrosa, K. Bloern, Y. van Kooyk and J. Rojo, Biomacromolecules, 2012, 13, 3209–3219 CrossRef CAS PubMed.
- J. J. Reina, S. Sattin, D. Invernizzi, S. Mari, L. Martinez-Prats, G. Tabarani, F. Fieschi, R. Delgado, P. M. Nieto, J. Rojo and A. Bernardi, ChemMedChem, 2007, 2, 1030–1036 CrossRef CAS PubMed.
- S. Maria, I. Sanchez-Medina, P. Mereghetti, L. Belvisi, J. Jimenez-Barbero and A. Bernardi, Carbohydr. Res., 2007, 342, 1859–1868 CrossRef PubMed.
- S. Mari, H. Posteri, G. Marcou, D. Potenza, F. Micheli, F. J. Canada, J. Jimenez-Barbero and A. Bernardi, Eur. J. Org. Chem., 2004, 5119–5125 CrossRef CAS.
- J. Luczkowiak, S. Sattin, I. Sutkeviciute, J. J. Reina, M. Sanchez-Navarro, M. Thepaut, L. Martinez-Prats, A. Daghetti, F. Fieschi, R. Delgado, A. Bernardi and J. Rojo, Bioconjugate Chem., 2011, 22, 1354–1365 CrossRef CAS PubMed.
- S. Sattin, A. Daghetti, M. Thepaut, A. Berzi, M. Sanchez-Navarro, G. Tabarani, J. Rojo, F. Fieschi, M. Clerici and A. Bernardi, ACS Chem. Biol., 2010, 5, 301–312 CrossRef CAS PubMed.
- A. Berzi, J. J. Reina, R. Ottria, I. Sutkeviciute, P. Antonazzo, M. Sanchez-Navarro, E. Chabrol, M. Biasin, D. Trabattoni, I. Cetin, J. Rojo, F. Fieschi, A. Bernardi and M. Clerici, AIDS, 2012, 26, 127–137 CrossRef CAS PubMed.
- N. Varga, I. Sutkeviciute, R. Ribeiro-Viana, A. Berzi, R. Ramdasi, A. Daghetti, G. Vettoretti, A. Amara, M. Clerici, J. Rojo, F. Fieschi and A. Bernardi, Biomaterials, 2014, 35, 4175–4184 CrossRef CAS PubMed.
- B. Bertolotti, I. Sutkeviciute, M. Ambrosini, R. Ribeiro-Viana, J. Rojo, F. Fieschi, H. Dvorakova, M. Kasakova, K. Parkan, M. Hlavackova, K. Novakova and J. Moravcova, Org. Biomol. Chem., 2017, 15, 3995–4004 RSC.
- J. J. Garcia-Vallejo, N. Koning, M. Ambrosini, H. Kalay, I. Vuist, R. Sarrami-Forooshani, T. B. H. Geijtenbeek and Y. van Kooyk, Int. Immunol., 2013, 25, 221–233 CrossRef CAS PubMed.
- S. Ordanini, N. Varga, V. Porkolab, M. Thepaut, L. Belvisi, A. Bertaglia, A. Palmioli, A. Berzi, D. Trabattoni, M. Clerici, F. Fieschi and A. Bernardi, Chem. Commun., 2015, 51, 3816–3819 RSC.
- H. C. Wen, C. H. Lin, J. S. Huang, C. L. Tsai, T. F. Chen and S. K. Wang, Chem. Commun., 2019, 55, 9124–9127 RSC.
- S. K. Wang, P. H. Liang, R. D. Astronomo, T. L. Hsu, S. L. Hsieh, D. R. Burton and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3690–3695 CrossRef CAS PubMed.
- J. Ramos-Soriano, M. C. de la Fuente, N. de la Cruz, R. C. Figueiredo, J. Rojo and J. J. Reina, Org. Biomol. Chem., 2017, 15, 8877–8882 RSC.
- N. de la Cruz, J. Ramos-Soriano, J. J. Reina, J. L. de Paz, M. Thepaut, F. Fieschi, A. Sousa-Herves and J. Rojo, Org. Biomol. Chem., 2020, 18, 6086–6094 RSC.
- S. J. Zhou, P. Trochimczyk, L. L. Sun, S. Hou and H. G. Li, Curr. Org. Chem., 2016, 20, 1490–1501 CrossRef CAS.
- E. Castro, A. H. Garcia, G. Zavala and L. Echegoyen, J. Mater. Chem. B, 2017, 5, 6523–6535 RSC.
- A. Hirsch, Top. Curr. Chem., 1999, 199, 1–65 CrossRef CAS.
- B. M. Illescas, J. Rojo, R. Delgado and N. Martín, J. Am. Chem. Soc., 2017, 139, 6018–6025 CrossRef CAS PubMed.
- H. Isobe, K. Cho, N. Solin, D. B. Werz, P. H. Seeberger and E. Nakamura, Org. Lett., 2007, 9, 4611–4614 CrossRef CAS PubMed.
- J. F. Nierengarten, J. Iehl, V. Oerthel, M. Holler, B. M. Illescas, A. Munoz, N. Martin, J. Rojo, M. Sanchez-Navarro, S. Cecioni, S. Vidal, K. Buffet, M. Durka and S. P. Vincent, Chem. Commun., 2010, 46, 3860–3862 RSC.
- Z. Shengju, T. Piotr, S. Lili, H. Sen and L. Hongguang, Curr. Org. Chem., 2016, 20, 1490–1501 CrossRef.
- I. Nierengarten and J.-F. Nierengarten, Chem. – Asian J., 2014, 9, 1436–1444 CrossRef CAS PubMed.
- J. F. Nierengarten, Chem. Commun., 2017, 53, 11855–11868 RSC.
- P. Compain, C. Decroocq, J. Iehl, M. Holler, D. Hazelard, T. M. Barragan, C. O. Mellet and J. F. Nierengarten, Angew. Chem., Int. Ed., 2010, 49, 5753–5756 CrossRef CAS PubMed.
- M. A. Flos, M. I. G. Moreno, C. O. Mellet, J. M. G. Fernandez, J. F. Nierengarten and S. P. Vincent, Chem. – Eur. J., 2016, 22, 11450–11460 CrossRef PubMed.
- F. Stauffert, A. Bodlenner, T. M. N. Trinh, M. I. Garcia-Moreno, C. O. Mellet, J. F. Nierengarten and P. Compain, New J. Chem., 2016, 40, 7421–7430 RSC.
- R. Risquez-Cuadro, J. M. G. Fernandez, J. F. Nierengarten and C. O. Mellet, Chem. – Eur. J., 2013, 19, 16791–16803 CrossRef CAS PubMed.
- A. Tikad, H. X. Fu, C. M. Sevrain, S. Laurent, J. F. Nierengarten and S. P. Vincent, Chem. – Eur. J., 2016, 22, 13147–13155 CrossRef CAS PubMed.
- M. Durka, K. Buffet, J. Iehl, M. Holler, J. F. Nierengarten and S. P. Vincent, Chem. – Eur. J., 2012, 18, 641–651 CrossRef CAS PubMed.
- K. Buffet, E. Gillon, M. Holler, J. F. Nierengarten, A. Imberty and S. P. Vincent, Org. Biomol. Chem., 2015, 13, 6482–6492 RSC.
- M. Durka, K. Buffet, J. Iehl, M. Holler, J. F. Nierengarten, J. Taganna, J. Bouckaert and S. P. Vincent, Chem. Commun., 2011, 47, 1321–1323 RSC.
- S. Cecioni, V. Oerthel, J. Iehl, M. Holler, D. Goyard, J. P. Praly, A. Imberty, J. F. Nierengarten and S. Vidal, Chem. – Eur. J., 2011, 17, 3252–3261 CrossRef CAS PubMed.
- M. Serda, K. Malarz, A. Mrozek-Wilczkiewicz, M. Wojtyniak, R. Musioł and S. A. Curley, Sci. Rep., 2020, 10, 260 CrossRef CAS PubMed.
- M. Serda, M. J. Ware, J. M. Newton, S. Sachdeva, M. Krzykawska-Serda, L. Nguyen, J. Law, A. O. Anderson, S. A. Curley, L. J. Wilson and S. J. Corr, Nanomedicine, 2018, 13, 2981–2993 CrossRef CAS PubMed.
- M. Sanchez-Navarro, A. Munoz, B. M. Illescas, J. Rojo and N. Martin, Chem. – Eur. J., 2011, 17, 766–769 CrossRef CAS.
- J. Luczkowiak, A. Munoz, M. Sanchez-Navarro, R. Ribeiro-Viana, A. Ginieis, B. M. Illescas, N. Martin, R. Delgado and J. Rojo, Biomacromolecules, 2013, 14, 431–437 CrossRef CAS PubMed.
- A. Munoz, D. Sigwalt, B. M. Illescas, J. Luczkowiak, L. Rodriguez-Perez, I. Nierengarten, M. Holler, J. S. Remy, K. Buffet, S. P. Vincent, J. Rojo, R. Delgado, J. F. Nierengarten and N. Martin, Nat. Chem., 2016, 8, 50–57 CrossRef CAS PubMed.
- H. P. Li, S. A. Haque, A. Kitaygorodskiy, M. J. Meziani, M. Torres-Castillo and Y. P. Sun, Org. Lett., 2006, 8, 5641–5643 CrossRef CAS PubMed.
- O. Engstrom, A. Munoz, B. M. Illescas, N. Martin, R. Ribeiro-Viana, J. Rojo and G. Widmalm, Org. Biomol. Chem., 2015, 13, 8750–8755 RSC.
- R. Ribeiro-Viana, M. Sanchez-Navarro, J. Luczkowiak, J. R. Koeppe, R. Delgado, J. Rojo and B. G. Davis, Nat. Commun., 2012, 3, 1303 CrossRef PubMed.
- T. M. N. Trinh, M. Holler, J. P. Schneider, M. I. Garcia-Moreno, J. M. G. Fernandez, A. Bodlenner, P. Compain, C. O. Mellet and J. F. Nierengarten, J. Mater. Chem. B, 2017, 5, 6546–6556 RSC.
- J. F. Nierengarten, J. P. Schneider, T. M. N. Trinh, A. Joosten, M. Holler, M. L. Lepage, A. Bodlenner, M. I. Garcia-Moreno, C. O. Mellet and P. Compain, Chem. – Eur. J., 2018, 24, 2483–2492 CrossRef CAS PubMed.
- A. Munoz, B. M. Illescas, J. Luczkowiak, F. Lasala, R. Ribeiro-Viana, J. Rojo, R. Delgado and N. Martin, J. Mater. Chem. B, 2017, 5, 6566–6571 RSC.
- A. Munoz, B. M. Illescas, M. Sanchez-Navarro, J. Rojo and N. Martin, J. Am. Chem. Soc., 2011, 133, 16758–16761 CrossRef CAS PubMed.
- J. L. Delgado, M. A. Herranz and N. Martin, J. Mater. Chem., 2008, 18, 1417–1426 RSC.
- J. Bartelmess, S. J. Quinn and S. Giordani, Chem. Soc. Rev., 2015, 44, 4672–4698 RSC.
- F. R. Baptista, S. A. Belhout, S. Giordani and S. J. Quinn, Chem. Soc. Rev., 2015, 44, 4433–4453 RSC.
- H. Y. Liu, L. N. Zhang, M. Yan and J. H. Yu, J. Mater. Chem. B, 2017, 5, 6437–6450 RSC.
- O. S. Kwon, H. S. Song, T. H. Park and J. Jang, Chem. Rev., 2019, 119, 36–93 CrossRef CAS PubMed.
- A. Kasprzak and M. Poplawska, Chem. Commun., 2018, 54, 8547–8562 RSC.
- S. Priyadarsini, S. Mohanty, S. Mukherjee, S. Basu and M. Mishra, J. Nanostruct. Chem., 2018, 8, 123–137 CrossRef CAS.
- S. A. Hill, D. Benito-Alifonso, D. J. Morgan, S. A. Davis, M. Berry and M. C. Galan, Nanoscale, 2016, 8, 18630–18634 RSC.
- S. A. Hill, D. Benito-Alifonso, S. A. Davis, D. J. Morgan, M. Berry and M. C. Galan, Sci. Rep., 2018, 8, 12234 CrossRef PubMed.
- K. S. Ibrahim, Carbon Lett., 2013, 14, 131–144 CrossRef.
- N. Kong, M. R. Shimpi, O. Ramstrom and M. D. Yan, Carbohydr. Res., 2015, 405, 33–38 CrossRef CAS PubMed.
- B. Dinesh, A. Bianco and C. Menard-Moyon, Nanoscale, 2016, 8, 18596–18611 RSC.
- S. Mallakpour and S. Soltanian, RSC Adv., 2016, 6, 109916 RSC.
- R. Alshehri, A. M. Ilyas, A. Hasan, A. Arnaout, F. Ahmed and A. Memic, J. Med. Chem., 2016, 59, 8149–8167 CrossRef CAS PubMed.
- M. F. Zhang, T. Yamaguchi, S. Iijima and M. Yudasaka, Nanomedicine, 2013, 9, 657–664 CrossRef CAS PubMed.
- L. Rodriguez-Perez, J. Ramos-Soriano, A. Perez-Sanchez, B. M. Illescas, A. Munoz, J. Luczkowiak, F. Lasala, J. Rojo, R. Delgado and N. Martin, J. Am. Chem. Soc., 2018, 140, 9891–9898 CrossRef CAS PubMed.
- M. E. Ragoussi, S. Casado, R. Ribeiro-Viana, G. de la Torre, J. Rojo and T. Torres, Chem. Sci., 2013, 4, 4035–4041 RSC.
- B. K. Price and J. M. Tour, J. Am. Chem. Soc., 2006, 128, 12899–12904 CrossRef CAS PubMed.
- G. Pagona, N. Karousis and N. Tagmatarchis, Carbon, 2008, 46, 604–610 CrossRef CAS.
- J. Ramos-Soriano, J. J. Reina, B. M. Illescas, N. de la Cruz, L. Rodriguez-Perez, F. Lasala, J. Rojo, R. Delgado and N. Martin, J. Am. Chem. Soc., 2019, 141, 15403–15412 CrossRef CAS PubMed.
- H. Feinberg, R. Castelli, K. Drickamer, P. H. Seeberger and W. I. Weis, J. Biol. Chem., 2007, 282, 4202–4209 CrossRef CAS PubMed.
- M. Thepaut, C. Guzzi, I. Sutkeviciute, S. Sattin, R. Ribeiro-Viana, N. Varga, E. Chabrol, J. Rojo, A. Bernardi, J. Angulo, P. M. Nieto and F. Fieschi, J. Am. Chem. Soc., 2013, 135, 2518–2529 CrossRef CAS PubMed.
- J. J. Reina, A. Di Maio, J. Ramos-Soriano, R. C. Figueiredo and J. Rojo, Org. Biomol. Chem., 2016, 14, 2873–2882 RSC.
- C. Ornelas, J. Broichhagen and M. Weck, J. Am. Chem. Soc., 2010, 132, 3923–3931 CrossRef CAS PubMed.
- J. Ramos-Soriano, J. J. Reina, A. Perez-Sanchez, B. M. Illescas, J. Rojo and N. Martin, Chem. Commun., 2016, 52, 10544–10546 RSC.
- J. Ramos-Soriano, J. J. Reina, B. M. Illescas, J. Rojo and N. Martin, J. Org. Chem., 2018, 83, 1727–1736 CrossRef CAS PubMed.
- F. Compostella, O. Pitirollo, A. Silvestri and L. Polito, Beilstein J. Org. Chem., 2017, 13, 1008–1021 CrossRef PubMed.
- M. Marradi, M. Martin-Lomas and S. Penades, Adv. Carbohydr. Chem. Biochem., 2010, 64, 211–290 CrossRef CAS PubMed.
- S. Toraskar, M. Gade, S. Sangabathuni, H. V. Thulasiram and R. Kikkeri, ChemMedChem, 2017, 12, 1116–1124 CrossRef CAS PubMed.
- X. Le Guevel, M. P. Perrino, T. D. Fernandez, F. Palomares, M. J. Torres, M. Blanca, J. Rojo and C. Mayorga, ACS Appl. Mater. Interfaces, 2015, 7, 20945–20956 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, Chem. Commun., 1994, 801–802 RSC.
| This journal is © The Royal Society of Chemistry 2021 |