
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of bifunctional disiloxanes via subsequent hydrosilylation of alkenes and alkynes†‡
Jakub
Szyling
 ab,
Rafał
Januszewski
ab,
Kamila
Jankowska
ab,
Jędrzej
Walkowiak
a,
Ireneusz
Kownacki
ab and
Adrian
Franczyk
*a
ab,
Rafał
Januszewski
ab,
Kamila
Jankowska
ab,
Jędrzej
Walkowiak
a,
Ireneusz
Kownacki
ab and
Adrian
Franczyk
*a
aCenter for Advanced Technology, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 10, Poznań 61-614, Poland. E-mail: adrian.franczyk@amu.edu.pl
bFaculty of Chemistry, Adam Mickiewicz University in Poznań, Uniwersytetu Poznańskiego 8, Poznań 61-614, Poland
First published on 8th April 2021
Abstract
The first protocol for the synthesis of unsymmetrical bifunctional 1,1,3,3-tetramethyldisiloxane derivatives via subsequent hydrosilylation of alkenes and alkynes is presented. The methodology described has vast functional group tolerance and is extremely efficient towards the formation of novel disiloxane-based building blocks.
The transition metal (TM) catalyzed addition of Si–H bonds to unsaturated bonds is the most powerful and commonly used laboratorial and industrial method for the preparation of silicon-based compounds.1–4 With practically unlimited potential for designing the organosilicon product structures and thus their chemical, physical, and biological properties, hydrosilylation is widely used in the synthesis of natural products,5 anti-corrosive coatings,6 hybrid materials,7,8 electrolytes,9,10 or polymer fire retardants.11 Of particular interest regarding organosilicon compounds in terms of their modifiability, is 1,1,3,3-tetramethyldisiloxane (1). This inexpensive, low hazard, stable towards air and moisture, and commercially available hydrosiloxane derivative with low molecular weight, constitutes an attractive building block that has been applied in the reductive cleavage of the inert C–O bonds,12 the reduction of tertiary carboxamides to tertiary amines,13 and the reduction of secondary and tertiary phosphine oxides.14 Moreover, functional disiloxanes have been widely applied as liquid crystals,15 solid polymeric electrolytes,16 or coupling agents.17
The functionalization of 1 can be achieved by hydrolysis/co-condensation of monochlorosilanes,18 condensation of silanolates,19 or more preferably the hydrosilylation reaction, because of its simplicity, efficiency, and safety.20,21 This final method was recently used for mono- or bisfunctionalization of 1 by γ-methacryloxypropyl,22–24 epoxy,22,25,26 aminopropyl,27 or ethyleneoxide28 groups or boryl, germyl,29 or silyl moieties.30–33 The literature reports described above concern the hydrosilylation of unsaturated carbon–carbon double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) in vinyl or allyl groups. To the best of our knowledge, the hydrosilylation of carbon–carbon triple bonds (C
C) in vinyl or allyl groups. To the best of our knowledge, the hydrosilylation of carbon–carbon triple bonds (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) by 1 or its monofunctionalized derivatives remains unexplored. The addition of the Si–H bond to the CC in terminal or internal alkynes can lead to a mixture of regio- and stereoisomers. Therefore, selective formation of unsaturated organosilicon compounds, which are extremely valuable building blocks in organic synthesis, is of importance.
C) by 1 or its monofunctionalized derivatives remains unexplored. The addition of the Si–H bond to the CC in terminal or internal alkynes can lead to a mixture of regio- and stereoisomers. Therefore, selective formation of unsaturated organosilicon compounds, which are extremely valuable building blocks in organic synthesis, is of importance.
Herein, we report the highly efficient TM-catalyzed bisfunctionalization of 1via the subsequent hydrosilylation of alkenes and alkynes. The main goal of our study was to develop a simple, straightforward, and easy to scale-up protocol that leads to the unique disiloxanes containing a broad gamut of reactive groups capable of further transformations.
In the first stage of our study, selective monofunctionalization of 1 with a series of olefins was carried out. The hydrosilylation occurred in the presence of commercially available chloro(1,5-cyclooctadiene) rhodium(I) dimer ([RhCl(cod)]2). As shown in our previous studies,20,21,29 the application of this binuclear rhodium catalyst (unlike the commonly used Karstedt's (Pt2(dvs)3) or Wilkinson's catalysts) allowed a reduction of 1 over olefins with the retention of excellent selectivity towards β-isomers, and negligible formation of bisadducts if 4-fold excess of 1 was used. Thus, we examined six different allylic derivatives (2a–f) and one vinyl derivative (2g) (Scheme 1). For 2a and 2c–e, the earlier protection of hydroxyl or amine group(s) with chlorotrimethylsilane (TMSCl) was needed to avoid competitive O- or N-silylation. All products with TMS-protected functionalities were obtained with almost quantitative yields and fully characterized (see the ESI‡).
 | ||
| Scheme 1 Hydrosilylation of allyl (2a–f) and vinyl (2g) derivatives with 1,1,3,3-tetramethyldisiloxane (1) in the presence of [RhCl(cod)]2. | ||
Hydrosilylation of alkenes (2a–g) was performed under the optimized conditions, in toluene (1 M) at 60 °C under an air atmosphere, in the presence of [RhCl(cod)]2 (10−4 mol per Rh). The reactions with (allyloxy)trimethylsilane (2a), allyl-glycidyl ether (2b), and N-allyl-N,N-bis(trimethylsilyl)amine (2d) resulted in the desired products 3a, 3b, and 3d with very high yields. However, the formation of bisadducts or α-isomer in a very low quantity (<6%) was also determined by GC–MS analysis. Application of TMS-protected 3-(allyloxy)propane-1,2-diol (2c), eugenol-TMS (2e), allylpentafluorobeznene (2f) and 2-chloro(ethyl)vinyl ether (2g) smoothly led to β-isomers without formation of the by-products. It is worth emphasizing that the monofunctionalized compounds 3c and 3f are herein reported and fully characterized for the first time. In addition, 3a, despite previous reports,34 has been completely spectrally characterized for the first time. The presented above process has good tolerance towards various functional groups such as silyloxy, glycidyl, amine, aryl, and halogen, which can be easily modified in further transformations. Additionally, for the hydrosilylation of 3a with 4b, the gram-scale synthesis was performed leading to 5ab with high efficiency.
In the next step with the series of diverse disiloxanes 3a–g in hand, we examined the scope and limitations for the synthesis of their bifunctional, unsymmetrical derivatives via hydrosilylation of internal and terminal alkynes. The selective hydrosilylation of CC bonds allows the introduction of an alkenyl moiety into the compound structure, which makes them an attractive building block in e.g., addition or coupling reactions. On the other hand, the selective hydrosilylation of the CC bonds, especially terminal ones, is a challenging task due to the possible formation of stereo- and regioisomers.
Based on our experience in hydrometallation of the unsaturated CC bonds, for the Si–H addition to internal alkynes, the commercially available and commonly used Karstedt's catalyst was selected.35 For terminal alkynes, in situ generated catalytic system based on PtO2/Xphos was the most efficient.35,36 Due to the vast diversity of monofunctionalized 3a–g derivatives and alkynes 4a–k, hydrosilylation was performed at 100 °C for 18 hours with the equimolar ratio of the substrates, which were used as received from the suppliers, without any initial purification step.
To determine the influence of alkyne structure on the process efficiency, we chose derivative 3a and used it in the hydrosilylation with internal (4a–d) and terminal alkynes (4e–k) (Table 1). The application of symmetrical and aromatic alkynes (4a–b) smoothly led to (E)-isomers (5aa–ab) via cis-addition of Si–H to the CC bond, with 99% conversion of reagents (Table 1, entries 1 and 2). In the same manner, aliphatic 4-octyne (4c) was transformed into 5ac (Table 1, entry 3). The hydrosilylation of unsymmetrical internal 4-[(trimethylsilyl)ethynyl]-phenylboronic acid pinacol ester (4d) with 3a resulted in the selective formation of the (E)-isomer. 1D NOESY analysis revealed that the proton from SiH group is attached to the alkenyl carbon with SiMe3 group (Table 1, entry 4; ESI,‡ Fig. S44).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Alkyne 4 | Conv. of 3aa [%] | Selectivity 5 = 6/7 or 5/6/7a [%] | Isolated yield of 5 [%] | |||
| R′ | R′′ | ||||||
Reaction conditions: for entries 1–4, 6: [3a]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[4a–d, f]:[Pt2(dsv)3] = 1:1:10−4 of Pt, 100 °C, 18 h, toluene (0.05–0.1 M); for entries 5, 7–12: [3a]:[4e–k]:[XPhos]:[PtO2] = 1:1:2 × 10−2:10−2 of PtO2, 100 °C, 18 h THF (0.07 M).a Determined by GC–MS or NMR analyses.b Analysed without any purification step.c Filtration through a syringe filter (0.2 μm) without further purification. :[4a–d, f]:[Pt2(dsv)3] = 1:1:10−4 of Pt, 100 °C, 18 h, toluene (0.05–0.1 M); for entries 5, 7–12: [3a]:[4e–k]:[XPhos]:[PtO2] = 1:1:2 × 10−2:10−2 of PtO2, 100 °C, 18 h THF (0.07 M).a Determined by GC–MS or NMR analyses.b Analysed without any purification step.c Filtration through a syringe filter (0.2 μm) without further purification. |
|||||||
| 1 | a | Ph | Ph | 99 | 100/0 | aa | 81 |
| 2 | b | 4-BrPh | 4-BrPh | 99 | 100/0 | ab | 77 |
| 3 | c | C3H7 | C3H7 | 100 | 100/0 | ac | 96b |
| 4 | d | 4-BpinPh | SiMe3 | 99 | 100/0 | ad | 73 |
| 5 | e | c-C6H11 | H | 96 | 98/2/0 | ae | 82 |
| 6 | f | CMe2(OSiMe3) | H | 100 | 85/15/0 | af | — |
| 7 | 100 | 100/0/0 | af | 94c | |||
| 8 | g | Ph | H | 100 | 98/2/0 | ag | 93 |
| 9 | h | 4-BrPh | H | 91 | 96/4/0 | ah | 69 |
| 10 | i | 4-MeOPh | H | 99 | 99/1/0 | ai | 95c |
| 11 | j | 9-Phenanthryl | H | 94 | 90/10/0 | aj | 68 |
| 12 | k | 3-Thienyl | H | 91 | 99/1/0 | ak | 84 |

Next, terminal alkyne screening was carried out. It was initially based on the reaction conditions determined for internal alkynes. Application of Pt2(dvs)3 resulted in a mixture of regioisomers 5 and 6, even for sterically hindered [(1,1-dimethyl-2-propynyl)oxy]trimethylsilane (4f) (5af/6af = 85/15). Thus, relying on our previous studies and literature reports, the generated in situ PtO2/Xphos system was applied. It is known as the selective catalyst for the addition of the Si–H bond to the CCH bond. Indeed, when it was used in the hydrosilylation of 4f with 3a, the exclusive formation of 5af occurred (Table 1, entry 7). For the linear 1-heptyne (4e), a very high selectivity was noticed as well. However, traces of 6ae were formed. The same observations were carried out for the aromatic terminal alkynes such as phenylacetylene (4g) (Table 1, entry 8). The incorporation of a weakly electron-withdrawing atom, i.e. Br, to the phenyl ring had a negative influence on the 3a conversion and reaction selectivity (5ah/6ah = 96/4) (Table 1, entry 9).
In turn, a donating group (–OMe) ensures a very high conversion of 3a and negligible formation of isomeric by-products (Table 1, entry 10). Polycyclic aromatic and heterocyclic alkynes such as 9-ethynylphenanthrene (4j) and 3-ethynylthiophene (4k) can also be effectively used for the hydrosilylation of 3a, although under the applied reaction conditions, a complete conversion of 3a was not observed (Table 1, entries 11 and 12).
Encouraged by the results obtained for the hydrosilylation of 3a with internal and terminal alkynes, we decided to expand the library of bifunctional unsymmetrical substituted derivatives. We applied 2b–g in the hydrosilylation of selected alkynes. The substrate scope is summarized in Scheme 2. In general, monosubstituted 1,1,3,3-tetramethyldisiloxane bearing glycidyl ether (3b) selectively reacted with aromatic (4a–b) and alkyl (4c) internal alkynes, smoothly leading to (E)-isomers (5ba–bc), whereas with terminal 4f, β-(E)-isomer (5bf) was formed. The same observation was made for the internal (4a–c) and terminal (4f) alkynes when 3c containing two OSiMe3 groups and 3d bearing N(SiMe3)2 groups were used. Our protocol was also suitable for the derivatives with eugenol (3e) or pentafluorobenzene (3f) moieties. For both, excellent selectivity and high isolation yields (≥71%) were observed (5ea–ef, 5fb, 5fc). The hydrosilylation of 1-(2-(2-chloroethoxy)ethyl)-1,1,3,3-tetramethyldisiloxane (3g) with internal and terminal alkynes, gave also the desired products (5gb, 5gf) with a very high reaction efficiency.
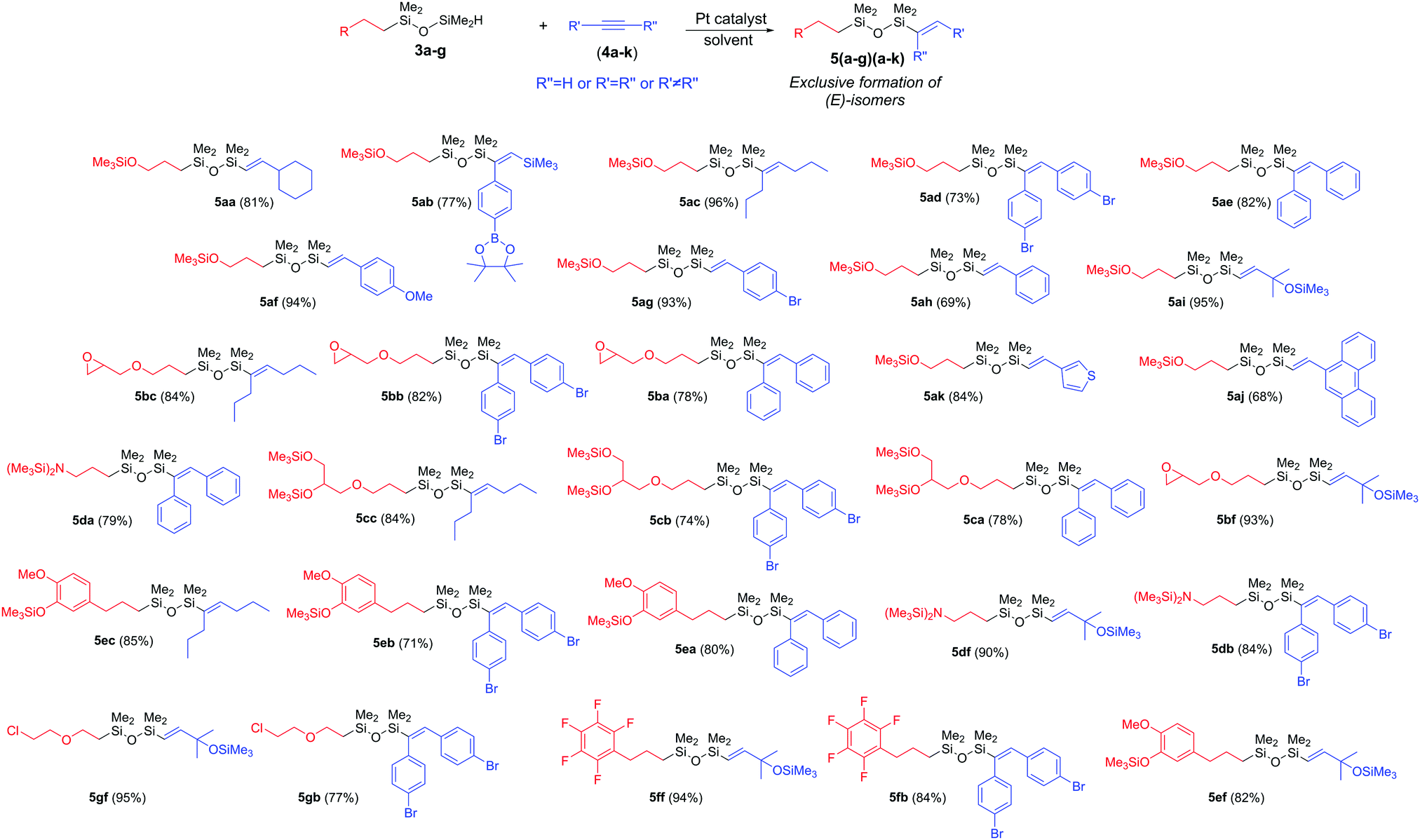 | ||
| Scheme 2 The scope of the synthesized bifunctional unsymmetrical substituted disiloxane derivatives. | ||
By the in situ FT-IR method, the influence of the structure of 3a–g on the rate of the hydrosilylation of diphenylacetylene (4a) was determined, by the following of the peak area at 907 cm−1 which corresponds to the Si–H bond. It was found that the addition of the Si–H bond to the CC proceeded rapidly within 1 hour for most siloxanes. Afterward, the reaction rate decreased due to the lower concentration of the substrates. The fastest consumption of siloxane occurred for 3a (2 h), while the slowest for 3b (over 7 h). For the rest of the reagents, the reaction time was between 3 and 6 hours (Fig. 1). For 3d and 3e, the peak conversion was not complete due to partial overlapping of the band assigned to Si–N and ether bonds with the Si–H band. However, GC analysis performed after flattening of the conversion curves confirmed the 100% consumption of the siloxanes.
 | ||
| Fig. 1 The hydrosilylation of 4a with 3a–g monitored by in situ FT-IR. | ||
It should be emphasized, that the presented protocol has excellent functional group tolerance for monosubstituted 1,1,3,3-tetramethyldisiloxanes (siloxy, glycidyl, amine, aryl, halogen groups) and alkynes (n-alkyl, c-alkyl, aryl, halogen, alkoxy, heterocyclic groups). Moreover, our approach is extremely selective towards the formation of (E)-isomers. The presence of the unsaturated CC bond on the one side and the reactive moiety on the other makes them very appealing building blocks with potential usage as coupling agents or in materials science. It is worth noting that all bifunctional 1,1,3,3-tetramethyldisiloxanes are new compounds and were comprehensively characterized by NMR, FT-IR, and MS methods. Moreover, a gram-scale reaction of 2a–c with 1 was carried out. Very high yields and selectivities of the monofunctionalized derivatives: 3a (93%), 3b (92%), and 3c (89%) were obtained.
In summary, selective and highly efficient methods for the synthesis of 29 unsymmetrical bifunctional derivatives via simple hydrosilylation of structurally different alkynes and alkenes with 1,1,3,3-tetramethyldisiloxane have been successfully developed. The process was highly selective when the hydrosilylation of alkenes was carried out in the presence of [RhCl(cod)]2 and for internal or terminal alkynes Pt2(dvs)3 or PtO2/XPhos were respectively used. The protocol is effective towards a broad spectrum of alkenes as well as internal and terminal alkynes with various functionalities, leading to products with high isolation yields. At the same time it is easy to scale up, and therefore the synthesis can be performed on the gram scale.
This work was supported by the National Centre for Research and Development in Poland, Lider Programme No. LIDER/6/0017/L-9/17/NCBR/2018 and the National Science Centre in Poland No. UMO-2019/32/C/ST4/00235, UMO-2018/31/G/ST4/04012.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Marciniec, Hydrosilylation: a comprehensive review on recent advances, Springer Science & Business Media, 2009 Search PubMed.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, 2017, pp. 569–620 Search PubMed.
- A. K. Roy, in Advances in Organometallic Chemistry, ed. R. West, A. F. Hill and M. J. Fink, Academic Press, 2007, vol. 55, pp. 1–59 Search PubMed.
- Y. Naganawa, K. Inomata, K. Sato and Y. Nakajima, Tetrahedron Lett., 2020, 61, 151513 CrossRef CAS.
- E. Langkopf and D. Schinzer, Chem. Rev., 1995, 95, 1375–1408 CrossRef CAS.
- J. Wojciechowski, K. Szubert, R. Peipmann, M. Fritz, U. Schmidt, A. Bund and G. Lota, Electrochim. Acta, 2016, 220, 1–10 CrossRef CAS.
- U. Díaz, T. García, A. Velty and A. Corma, Chem. – Eur. J., 2012, 18, 8659–8672 CrossRef PubMed.
- Y. Du and H. Liu, Dalton Trans., 2020, 49, 5396–5405 RSC.
- S. L. Guillot, A. Peña-Hueso, M. L. Usrey and R. J. Hamers, J. Electrochem. Soc., 2017, 164, A1907–A1917 CrossRef CAS.
- T. Yong, J. Wang, Y. Mai, X. Zhao, H. Luo and L. Zhang, J. Power Sources, 2014, 254, 29–32 CrossRef CAS.
- Z. Han, A. Fina and G. Camino, Polymer Green Flame Retardants, Elsevier, 2014, pp. 389–418 Search PubMed.
- P. Alvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352–17353 CrossRef CAS.
- Y. Sunada, H. Kawakami, T. Imaoka, Y. Motoyama and H. Nagashima, Angew. Chem., Int. Ed., 2009, 48, 9511–9514 CrossRef CAS.
- M. Berthod, A. Favre-Réguillon, J. Mohamad, G. Mignani, G. Docherty and M. Lemaire, Synlett, 2007, 1545–1548 CAS.
- G. Shanker, M. Prehm and C. Tschierske, J. Mater. Chem., 2012, 22, 168–174 RSC.
- I. J. Lee, G. S. Song, W. S. Lee and D. H. Suh, J. Power Sources, 2003, 114, 320–329 CrossRef CAS.
- F. J. LaRonde, A. M. Ragheb and M. A. Brook, Colloid Polym. Sci., 2003, 281, 391–400 CrossRef CAS.
- Y. Isoda and K. Ayama, EP0657486B1, 1998.
- H. Friedrich, I. Jansen and K. Rühlmann, Polym. Degrad. Stab., 1994, 46, 9–18 CrossRef CAS.
- R. Januszewski, I. Kownacki, H. Maciejewski and B. Marciniec, J. Organomet. Chem., 2017, 846, 263–268 CrossRef CAS.
- R. Januszewski, I. Kownacki, H. Maciejewski, B. Marciniec and A. Szymańska, Eur. J. Inorg. Chem., 2017, 851–856 CrossRef CAS.
- R. Acosta Ortiz, M. Sangermano, R. Bongiovanni, A. E. Garcia Valdez, L. B. Duarte, I. P. Saucedo and A. Priola, Prog. Org. Coat., 2006, 57, 159–164 CrossRef CAS.
- G. Erdodi and J. P. Kennedy, J. Polym. Sci., Part A-1: Polym. Chem., 2007, 45, 295–307 CrossRef CAS.
- G. Guzman, T. Nugay, I. Nugay, N. Nugay, J. Kennedy and M. Cakmak, Macromolecules, 2015, 48, 6251–6262 CrossRef CAS.
- R. Malik and J. V. Crivello, J. Macromol. Sci., Pure Appl. Chem., 1997, 34, 247–263 CrossRef.
- D. Zhang, E. Liang, T. Li, S. Chen, J. Zhang, X. Cheng, J. Zhou and A. Zhang, RSC Adv., 2013, 3, 3095–3102 RSC.
- Y.-Z. Niu, L. Zhang, S.-J. Liang, D.-X. Wang and S.-Y. Feng, Chin. Chem. Lett., 2014, 25, 1419–1422 CrossRef CAS.
- S. S. Sologubov, A. V. Markin, N. N. Smirnova, N. A. Novozhilova, E. A. Tatarinova and A. M. Muzafarov, J. Phys. Chem. B, 2015, 119, 14527–14535 CrossRef CAS PubMed.
- R. Januszewski, M. Grzelak, B. Orwat, M. Dutkiewicz and I. Kownacki, J. Catal., 2020, 390, 103–108 CrossRef CAS.
- D. A. de Vekki, V. Ol’Sheev, V. Spevak and N. Skvortsov, Russ. J. Gen. Chem., 2001, 71, 1912–1923 CrossRef CAS.
- D. A. de Vekki and N. Skvortsov, Russ. J. Gen. Chem., 2004, 74, 197–206 CrossRef CAS.
- D. A. de Vekki, V. Uvarov, A. Reznikov and N. Skvortsov, Russ. Chem. Bull., 2008, 57, 349–357 CrossRef.
- R. Murthy, C. D. Cox, M. S. Hahn and M. A. Grunlan, Biomacromolecules, 2007, 8, 3244–3252 CrossRef CAS PubMed.
- G. D. Khatuntsev, V. D. Sheludyakov and V. F. Mironov, Zh. Obshch. Khim., 1974, 43, 2150–2155 Search PubMed.
- K. Stefanowska, A. Franczyk, J. Szyling, K. Salamon, B. Marciniec and J. Walkowiak, J. Catal., 2017, 356, 206–213 CrossRef CAS.
- A. Hamze, O. Provot, J.-D. Brion and M. Alami, J. Organomet. Chem., 2008, 693, 2789–2797 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Bogdan Marciniec on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc01253c |
| This journal is © The Royal Society of Chemistry 2021 |