
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct synthesis of polyureas from the dehydrogenative coupling of diamines and methanol†
Amit
Kumar
 *a,
Daniel
Armstrong
a,
Gavin
Peters
a,
Manjula
Nagala
b and
Sally
Shirran
b
*a,
Daniel
Armstrong
a,
Gavin
Peters
a,
Manjula
Nagala
b and
Sally
Shirran
b
aSchool of Chemistry, University of St. Andrews, North Haugh, St. Andrews, KY169ST, UK. E-mail: ak336@st-andrews.ac.uk
bBSRC Mass Spectrometry and Proteomics Facility, University of St. Andrews, North Haugh, St. Andrews, KY169ST, UK
First published on 21st May 2021
Abstract
We report here the first example of the direct synthesis of polyureas from the dehydrogenative coupling of diamines and methanol using a ruthenium pincer catalyst. The present methodology replaces the use of toxic diisocyanates, conventionally used for the production of polyureas, with methanol, which is renewable, less toxic, and cheaper, making the overall process safer and more sustainable. Further advantages of the current method have been demonstrated by the synthesis of a renewable, a chiral, and the first 13C-labelled polyurea.
Polyureas are common plastics with substantial applications in coating, adhesive and biomedical materials. Their current annual global market is worth USD 885 million and is expected to reach USD 1481 million by 2025.1 The current state-of-the-art technology for the production of polyureas involves the reaction of diamines with diisocyanates. However, diisocyanates and their precursors (phosgene gas) are extremely toxic. Two approaches towards diisocyanate-free synthesis of polyureas have been reported. The first one is based on the synthesis of polyureas from the polycondensation of diamines with CO2.2–7 Although the use of CO2 for the synthesis of polyureas is attractive, the use of harsh reaction conditions such as reaction temperature of 170–180 °C, and CO2 pressure of 40–110 bars, in order to overcome the high thermodynamic barriers, present a bottleneck to utilize this methodology on an industrial level. Another approach is based on the polycondensation of diamines with CO2 derivatives – organic carbamates,8–12 organic carbonates13–15 and urea (NH2CONH2).16,17 This methodology also suffers from drawbacks such as the use of expensive reagents or solvents (e.g. biscarbamate8–12 or ionic liquids as solvent), poor substrate scope,14–17 and low atom-economy.13
Catalytic dehydrogenation is a green and atom-economic approach in organic synthesis, and several new green protocols based on acceptorless dehydrogenative coupling have been developed in the past.18 For example, in relevance to the current report, the synthesis of urea derivatives has been reported by the catalytic dehydrogenative coupling of methanol and amines, independently by the groups of Hong,19 Gunanathan,20 Bernskoetter,21 and Milstein.22 The concept of acceptorless dehydrogenative coupling has also been employed for the synthesis of useful polymers such as polyesters,23 and nylons.24,25 Prakash and co-workers speculated that a white precipitate formed during the dehydrogenative synthesis of cyclic urea could be a polyurea.26 However, it was not confirmed by any characterization details. To the best of our knowledge, dehydrogenative synthesis of polyureas has not been reported before. In light of the recent developments on the synthesis of urea derivatives from amines and methanol, and the urgent need of chemical industries to shift towards a circular economy,27 we envisioned the synthesis of a class of useful plastic - polyureas by the catalytic dehydrogenative coupling of diamines and methanol (Scheme 1).
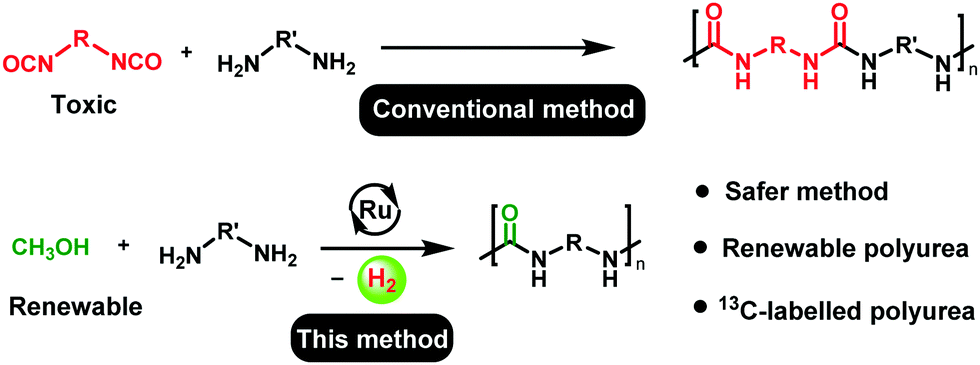 | ||
| Scheme 1 Synthesis of polyureas using the conventional method versus the method disclosed herein. | ||
The unique advantage of the discovered methodology is the use of methanol, which can be produced from biomass or by the direct hydrogenation of CO2.28,29 The concept of utilizing methanol for the purpose of fuel and materials has been advocated by Olah and Prakash as a so-called “methanol economy”.30 Expanding on the vision of the methanol economy, we also present here the synthesis of a new renewable polyurea from the dehydrogenative coupling of a bio-sourced diamine with methanol that can be renewable. Furthermore, using 13CH3OH we have also demonstrated the first synthesis of 13C-labelled polyureas which may have potential applications in drug delivery,31–3313C-MRI,34 and in assessing the environmental impacts of plastics.35
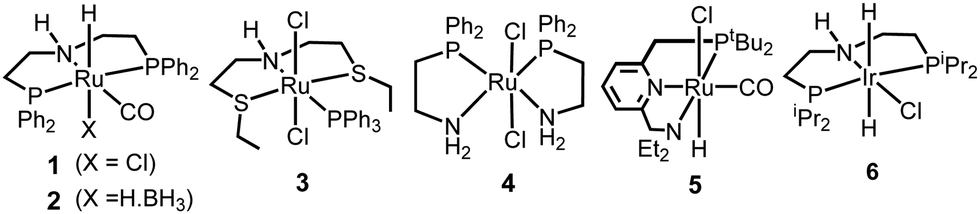
We started our investigation by exploring optimum conditions for the dehydrogenative coupling of diamines and methanol in the presence of ruthenium-Macho catalyst 1 which in the past has exhibited excellent activity for dehydrogenation of methanol36 and the dehydrogenative synthesis of urea derivatives.19 Refluxing a toluene solution of 4,7,10-trioxa-1,13-tridecanediamine (2 mmol) and methanol (4 mmol) in the presence of complex 1 (1 mol%) and KOtBu (2 mol%) under a flow of nitrogen gas (open system, 24 h, 130 °C) did not result in the formation of any polyurea, presumably due to loss of methanol under the reaction conditions. Interestingly, when the same reaction was performed in a closed Young's flask, a white solid was isolated in 95% yield (Table 1, entry 1). H2 gas was confirmed by analysing the gas present in the headspace by the GC. An IR spectrum of the white solid showed a strong band at 1612 cm−1 characteristic of a carbonyl stretching frequency. Signals corresponding to the N–H stretching and bending were observed at 3319 and 1570 cm−1. These signals are in good agreement with the IR signals corresponding to a polyurea made from the reaction of CO2 with the analogous diamine suggesting that the white solid isolated in this case is a polyurea A.2,6 This was further confirmed by a MALDI-TOF mass spectrometry studies that showed successive oligomeric signals corresponding to the polyurea A. 1H and 13C{1H} NMR spectra also corroborated the formation of polymer A. End group analysis by the 1H NMR spectroscopy showed the average molecular weight (Mn) of the polymer to be 4060 Da. Having confirmed the identity of the polymer, we studied the effects of catalytic conditions on the yield and Mn of the polymer. Lower values of Mn were obtained at lower reaction times (6, and 12 h), whereas increasing the reaction time to 48 h did not significantly change the value of Mn (4300 Da, see ESI†). Moreover, using 1 mol% KOtBu while keeping the remaining conditions same, resulted in a lower yield and Mn of the polymer (Table 1, entry 2), suggesting that the base is involved in assisting the dehydrogenation process in addition to generating the active species (by deprotonation of the N–H proton and removal of the chloride ligand in 1). The presence of excess base has been previously found to facilitate the dehydrogenation of alcohol by promoting the C–H cleavage step37 or by scavenging impurities.38 Performing a controlled experiment using the purified (distilled and degassed) diamine resulted in only 3% increment in the yield in comparison to that of entry 2 suggesting the important role of the base in the catalytic cycle. Under the analogous conditions as described in Table 1, catalysts 2–6 exhibited poor to no activity (entries 3–8). Furthermore, when the reaction was performed in the presence of the Ru(PPh3)3HClCO, which is a precursor to the synthesis of complex 1, no formation of polyurea was observed, suggestive of the important role of the PNP pincer ligand. Low polymer yields of 15% and 25% were obtained in the cases of anisole and DMSO solvents, respectively (entries 9–10). However, in the case of THF, a relatively high yield of polymer (85%) was obtained (entry 11). Interestingly, the molecular weight of the polymer formed in the case of THF was found to be higher than that formed using toluene. Performing the catalysis under a neat condition in the absence of a solvent resulted in a low yield (25%) of the polymer (entry 12). Furthermore, using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of diamine and methanol also resulted in a lower yield (22%) of polyurea (entry 13).
:1 ratio of diamine and methanol also resulted in a lower yield (22%) of polyurea (entry 13).
|
|
||||
|---|---|---|---|---|
| Entry | Precatalyst | Solvent | Yield (%) | Mol Wt (Mn) |
| a Catalytic conditions: 4,7,10-trioxa-1,13-tridecanediamine (440 mg, 2 mmol), methanol (3.2 mL, 8 mmol), precatalyst (1–6, 0.02 mmol), KOtBu (4.5 mg, 0.04 mmol), solvent (2 mL), 130 °C, 24 h. b 0.02 mmol of KOtBu was used. c No KOtBu was used. d 2 mmol of methanol was used. Mn is in Dalton (Da). | ||||
| 1 | 1 | Toluene | 95 | 4060 |
| 2b | 1 | Toluene | 55 | 2100 |
| 3b | 2 | Toluene | 32 | 1500 |
| 4c | 2 | Toluene | 10 | — |
| 5 | 3 | Toluene | 8 | — |
| 6 | 4 | Toluene | 0 | — |
| 7 | 5 | Toluene | 24 | 900 |
| 8 | 6 | Toluene | 34 | 800 |
| 9 | 1 | Anisole | 15 | 2500 |
| 10 | 1 | DMSO | 25 | 1500 |
| 11 | 1 | THF | 85 | 4757 |
| 12 | 1 | Neat | 25 | — |
| 13d | 1 | Toluene | 22 | — |
|
||||
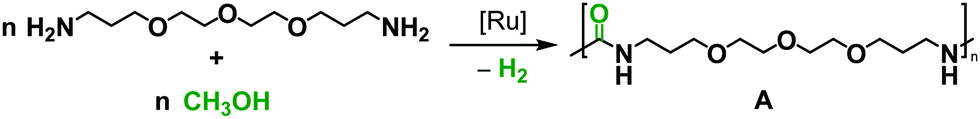
Under the optimized catalytic conditions, we synthesized a broad variety of polyureas (Table 2). Except for the polyurea A (Table 2, entry 1) which was soluble in H2O, CHCl3, DMF, and DMSO, all of the remaining polyureas were either insoluble or exhibited a very poor solubility in common solvents such as toluene, CHCl3, H2O, THF, acetone, DMF and DMSO (<10 mg mL−1). Therefore, we performed the molecular weight analysis by the 1H NMR spectroscopy and MALDI-TOF mass spectrometry in (deuterated) trifluoroacetic acid solvent. As described in Table 2, excellent yields of polyureas constituting the alkyl and aromatic groups were obtained with the Mn range of 2214–5500 Da. PDI were found to be in the range of 1.03–1.32 as estimated by the MALDI-TOF mass spectrometry (see ESI†). Decomposition temperatures (Td), as recorded by the thermal gravimetric analysis (TGA) at the 5% weight loss, were found to be in the range of 230 °C–299 °C. Melting temperatures (Tm) and glass transition temperatures (Tg) were recorded by the differential scanning calorimetry (DSC) measurements and found to vary with the diamines as mentioned in Table 2.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Diamine | Yield (%) | M n | T d (°C) | T m (°C) | T g (°C) |
| a Catalytic conditions: diamine (2 mmol), methanol (3.2 mL, 8 mmol), complex 1 (12 mg, 0.02 mmol), KOtBu (4.5 mg, 0.04 mmol), THF (2 mL), 130 °C, 24 h. b Toluene was used as a solvent. Mol wt (Mn) is in Da. | ||||||
| 1 |
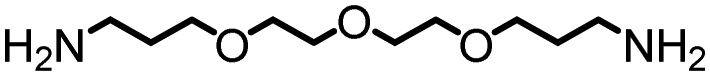
|
85 | 4757 | 280 | NA | NA |
| 2 |
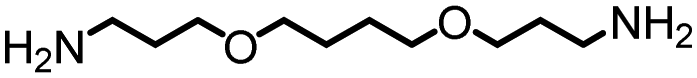
|
80 | 5500 | 290 | 119 | NA |
| 3b |
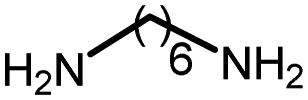
|
92 | 3173 | 230 | NA | 24 |
| 4b |
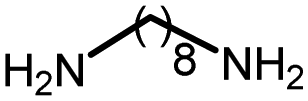
|
95 | 4063 | 237 | 241 | NA |
| 5b |
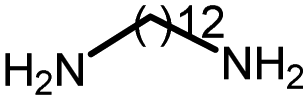
|
96 | 3476 | 258 | 208 | NA |
| 6 |
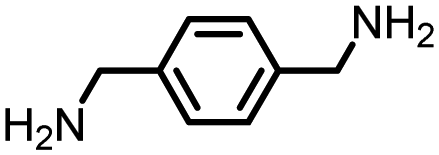
|
97 | 2592 | 299 | NA | −42 |
| 7 |
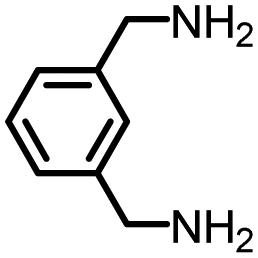
|
88 | 2214 | 259 | NA | −24 |
| 8 |
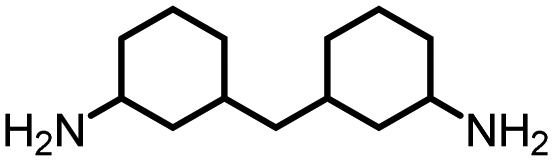
|
86 | 4498 | 290 | NA | NA |
| 9b |
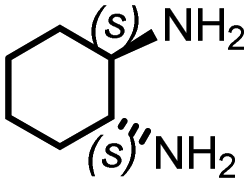
|
80 | 2862 | 289 | NA | NA |
| 10b |
|
75 | 2763 | 230 | NA | NA |
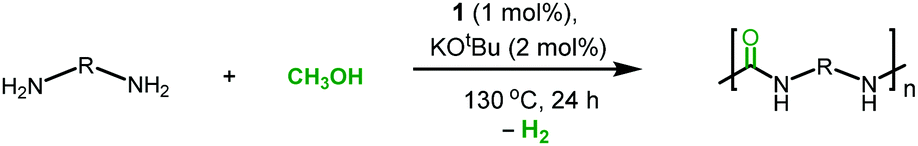
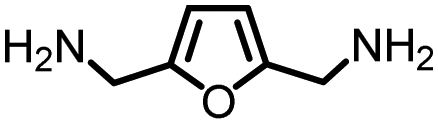
Furthermore, as 100% renewable methanol is industrially available,29 we envisioned to utilize this approach for the synthesis of renewable polyureas. We utilized 2,5-bis-(aminomethyl)furan (BAMF, Table 2, entry 10) which can be synthesized by amination of 2,5-diformylfuran obtained from biomass as patented by Evonik.39 Interestingly, a glassy solid characterised to be a polyurea was isolated in the 75% yield under the conditions described in Table 2. Moreover, the synthesis of m-xylenediamine has also been demonstrated from biomass making the corresponding polymer a potentially renewable polyurea (Table 2, entry 7).40
Chiral polyureas have been utilized for optical resolution,41 asymmetric catalysis,42 and recently for conformational deracemization of liquid crystals.43 Realizing the potential applications of chiral polyureas, we utilized this catalytic methodology for the synthesis of a chiral polyurea from the inexpensive and commercially available (1S,2S)-1,2-diaminocyclohexane. The corresponding polyurea was isolated in 80% yield (Table 2, entry 9). Its optical rotation was found to be [α]20D = −24.4. Interestingly, the sign of rotation was found to be opposite to that of the monomer whose optical rotation was found to be [α]20D = +20.0 as also reported earlier.42 No optical rotation was observed in the case of the polymer made from the racemic 1,2-diaminocyclohexane confirming that chirality of the polyurea comes from the use of a chiral monomer and not because of a helical arrangement.
Furthermore, we envisioned that the current methodology could allow us to synthesize the first 13C-labelled polyurea using 13CH3OH. Polyureas have potential applications as drug carriers32 for drug delivery, and a labelled polymer can offer a new tool to study the fate of such drug carriers.31,33 Moreover, 13C-labelled polymers have been utilized for 13C-MRI,34 and to study the environmental impact of microplastics.35 It is noteworthy that synthesis of a 13C-labelled polyurea from the previously reported methods would be very challenging and expensive due to the unavailability of a 13C-labelled diisocyanate. Using 4,7,10-trioxa-1,13-tridecanediamine and 13CH3OH under the conditions described in Table 2, we synthesized the first 13C-labelled polyurea 13C-A in 92% yield. The carbonyl frequency in the IR spectrum of 13C-A was found to decrease by 42 cm−1 in comparison to that of A confirming the 13C-labelling of the carbonyl carbon (see ESI,† Section 12).44 The isotope labelling was further confirmed by the MALDI-TOF mass spectrometry which showed a repetitive mass difference of 247 Da, 1 Da higher than that of the unlabelled polymer A (see ESI,† Section 12), and the 13C{1H} NMR spectroscopy.
Mechanisms for the dehydrogenative coupling of methanol and amines to form urea derivatives using ruthenium pincer catalysts have been proposed earlier based on the experimental and DFT studies.19,21,22 We suggest that the dehydrogenative synthesis of polyureas follows the analogous pathway as outlined in Scheme 2. The active species, that is generated by the reaction of complex 1 with KOtBu, dehydrogenates CH3OH to HCHO. This step has been previously studied as a part of the aqueous reforming of methanol.45 HCHO reacts with the nucleophilic amine group of a diamine to form a formamide and H2via a hemiaminal intermediate. The presence of oligomers containing formamide end group has been detected by the MALDI-TOF mass spectrometry and NMR spectroscopy (see ESI†). Formamide further reacts with the amine group of another diamine molecule to form a urea derivative and H2 as recently reported by Gunanathan20 and Milstein.22
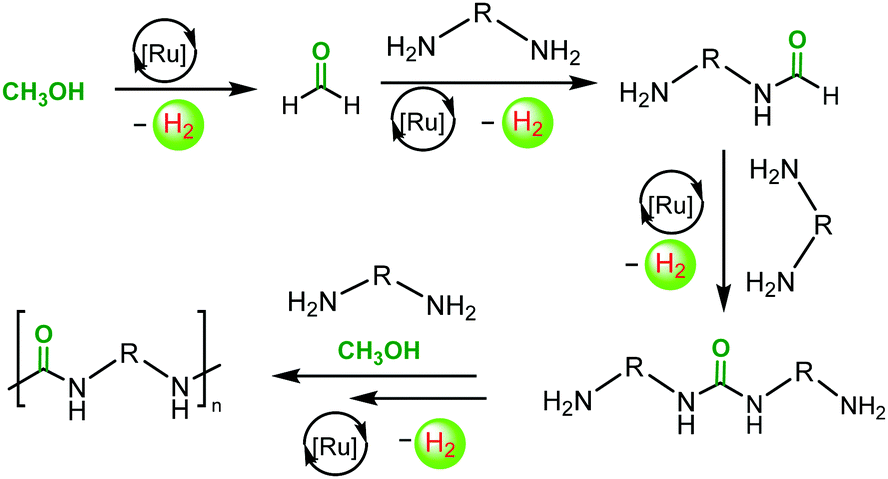 | ||
| Scheme 2 Proposed pathway for the synthesis of a polyurea from the dehydrogenative coupling of diamines and methanol. | ||
In conclusion, we present here the first example of the synthesis of a range of polyureas from the coupling of diamines and methanol. Reactions are catalysed by a ruthenium pincer complex and liberate H2 gas, which is valuable by itself, as the only by-product making the process highly atom economic. The present methodology has several advantages over the current state-of-the-art technology, for example, (a) it substitutes the use of toxic diisocyanates with methanol, which is less toxic and cheaper, making the process safer for the environment and human health, (b) it allows the synthesis of 100% renewable polyureas, and (c) it allows the unprecedented access to new 13C-labelled polyureas opening new windows of innovation in biomedical and material industry.
AK thanks the Leverhulme Trust for an early career fellowship. We thank the research group of Prof. Matt Clarke (School of Chemistry, University of St. Andrews) for assisting with lab equipment/facilities. We also thank Dr Robert Tooze and Mr Brian Boardman from the Drochaid Research Services Ltd for their help with the GC.
Conflicts of interest
The authors declare no conflict of interest.References
- Polyurea Coatings Market Global Forecast to 2025 |MarketsandMarkets, https://www.marketsandmarkets.com/.
- S. Jiang, R. Shi, H. Cheng, C. Zhang and F. Zhao, Green Energy Environ., 2017, 2, 370–376 CrossRef.
- P. Wang, X. Ma, Q. Li, B. Yang, J. Shang and Y. Deng, RSC Adv., 2016, 6, 54013–54019 RSC.
- P. Wang, Y. Fei, Y. Long and Y. Deng, J. CO2 Util., 2018, 28, 403–407 CrossRef CAS.
- P. Wang, Y. Fei and Y. Deng, New J. Chem., 2018, 42, 1202–1207 RSC.
- S. Jiang, H. Y. Cheng, R. H. Shi, P. X. Wu, W. W. Lin, C. Zhang, M. Arai and F. Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 47413–47421 CrossRef CAS PubMed.
- P. X. Wu, H. Y. Cheng, R. H. Shi, S. Jiang, Q. F. Wu, C. Zhang, M. Arai and F. Y. Zhao, Adv. Synth. Catal., 2019, 361, 317–325 CrossRef CAS.
- N. Kébir, M. Benoit, C. Legrand and F. Burel, Eur. Polym. J., 2017, 96, 87–96 CrossRef.
- A. Martin, L. Lecamp, H. Labib, F. Aloui, N. Kébir and F. Burel, Eur. Polym. J., 2016, 84, 828–836 CrossRef CAS.
- S. Ma, E. P. A. van Heeswijk, B. A. J. Noordover, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, ChemSusChem, 2018, 11, 149–158 CrossRef CAS PubMed.
- S. Ma, C. Liu, R. J. Sablong, B. A. J. Noordover, E. J. M. Hensen, R. A. T. M. Van Benthem and C. E. Koning, ACS Catal., 2016, 6, 6883–6891 CrossRef CAS.
- H. Y. Chen, W. C. Pan, C. H. Lin, C. Y. Huang and S. A. Dai, J. Polym. Res., 2012, 19, 9754–9758 CrossRef.
- M. Soccio, R. Mazzoni, C. Lucarelli, S. Quattrosoldi, A. Cingolani, M. Fiorini, N. Lotti and T. Tabanelli, ACS Sustainable Chem. Eng., 2020, 8, 15640–15650 CrossRef CAS.
- A. K. Qaroush, A. S. Al-Hamayda, Y. K. Khashman, S. I. Vagin, C. Troll and B. Rieger, Catal. Sci. Technol., 2013, 3, 2221–2226 RSC.
- W. C. Pan, K. Liao, C. H. Lin and S. A. Dai, J. Polym. Res., 2015, 22, 114 CrossRef.
- J. M. Dennis, L. I. Steinberg, A. M. Pekkanen, J. Maiz, M. Hegde, A. J. Müller and T. E. Long, Green Chem., 2018, 20, 243–249 RSC.
- J. M. Sirrine, S. A. Schexnayder, J. M. Dennis and T. E. Long, Polymer, 2018, 154, 225–232 CrossRef CAS.
- A. Kumar and C. Gao, ChemCatChem, 2021, 13, 1105–1134 CrossRef CAS.
- S. H. Kim and S. H. Hong, Org. Lett., 2016, 18, 212–215 CrossRef CAS PubMed.
- V. Krishnakumar, B. Chatterjee and C. Gunanathan, Inorg. Chem., 2017, 56, 7278–7284 CrossRef CAS PubMed.
- E. M. Lane, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2018, 9, 4003–4008 RSC.
- J. Bruffaerts, N. Von Wolff, Y. Diskin-Posner, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2019, 141, 16486–16493 CrossRef CAS PubMed.
- D. M. Hunsicker, B. C. Dauphinais, S. P. Mc Ilrath and N. J. Robertson, Macromol. Rapid Commun., 2012, 33, 232–236 CrossRef CAS PubMed.
- B. Gnanaprakasam, E. Balaraman, C. Gunanathan and D. Milstein, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1755–1765 CrossRef CAS.
- H. Zeng and Z. Guan, J. Am. Chem. Soc., 2011, 133, 1159–1161 CrossRef CAS PubMed.
- J. Kothandaraman, S. Kar, R. Sen, A. Goeppert, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2017, 139, 2549–2552 CrossRef CAS PubMed.
- T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS PubMed.
- Q. I. Roode-Gutzmer, D. Kaiser and M. Bertau, ChemBioEng Rev., 2019, 6, 209–236 CrossRef CAS.
- CRI – Carbon Recycling International, https://www.carbonrecycling.is/.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley, 2009 Search PubMed.
- R. C. A. Schellekens, F. Stellaard, H. J. Woerdenbag, H. W. Frijlink and J. G. W. Kosterink, Br. J. Clin. Pharmacol., 2011, 72, 879–897 CrossRef CAS PubMed.
- P. Rocas, C. Cusco, J. Rocas and F. Albericio, Curr. Drug Delivery, 2018, 15, 37–43 CAS.
- L. Plapied, N. Duhem, A. des Rieux and V. Préat, Curr. Opin. Colloid Interface Sci., 2011, 16, 228–237 CrossRef CAS.
- H. Yamada, Y. Hasegawa, H. Imai, Y. Takayama, F. Sugihara, T. Matsuda, H. Tochio, M. Shirakawa, S. Sando, Y. Kimura, A. Toshimitsu, Y. Aoyama and T. Kondo, J. Am. Chem. Soc., 2015, 137, 799–806 CrossRef CAS PubMed.
- M. Sander, H. P. E. Kohler and K. McNeill, Nat. Nanotechnol., 2019, 14, 301–303 CrossRef CAS PubMed.
- M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- E. Alberico, A. J. J. Lennox, L. K. Vogt, H. Jiao, W. Baumann, H.-J. Drexler, M. Nielsen, A. Spannenberg, M. P. Checinski, H. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 14890–14904 CrossRef CAS.
- D. H. Nguyen, X. Trivelli, F. Capet, Y. Swesi, A. FavreRéguollon, L. Vanoye, F. Dumeignil and R. M. Gauvin, ACS Catal., 2018, 8, 4719–4734 CrossRef CAS.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfu, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- I. Scodeller, S. Mansouri, D. Morvan, E. Muller, K. de Oliveira Vigier, R. Wischert and F. Jérôme, Angew. Chem., Int. Ed., 2018, 57, 10510–10514 CrossRef CAS PubMed.
- M. Hatanaka, Y. Nishioka and M. Yoshikawa, J. Membr. Sep. Technol., 2013, 2, 109–119 CAS.
- B. Dunjic, P. Gamez, F. Fache and M. Lemaire, J. Appl. Polym. Sci., 1996, 59, 1255–1262 CrossRef CAS.
- A. Zoabi, M. G. Santiago, D. Gelman, C. Rosenblatt, D. Avnir and R. Abu-Reziq, J. Phys. Chem. C, 2018, 122, 17936–17941 CrossRef CAS.
- G. John Karabatsos, J. Org. Chem., 1960, 25, 315–318 CrossRef.
- J. Kothandaraman, S. Kar, A. Goeppert, R. Sen and G. K. S. Prakash, Top. Catal., 2018, 61, 542–559 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc01121a |
| This journal is © The Royal Society of Chemistry 2021 |