
Light actuated stable radicals of the 9-anthracene carboxylic acid for designing new photochromic complexes†
Qi
Li
,
Qian
Zhang
,
Wu-Ji
Wei
,
A-Ni
Wang
,
Ji-Xiang
Hu
 * and
Guo-Ming
Wang
* and
Guo-Ming
Wang
College of Chemistry and Chemical Engineering, Qingdao University, Shandong 266071, P. R. China. E-mail: hujixiang@qdu.edu.cn
First published on 18th March 2021
Abstract
The photogeneration of stable radicals is important but still challenging in the field of optical switching, displays, and other devices. Herein, crystalline 9-anthracene carboxylic acid (9-AC) and a mononuclear complex constructed from this ligand were for the first time discovered to show radical-induced photochromism and photomagnetism after Xe lamp light irradiation. This study finds a simple radical-actuated photochromic molecule for constructing a novel system of photochromic materials.
9-Anthracene carboxylic acid (9-AC), as a rigid organic molecule, has been commonly utilized in the construction of coordination complexes.1–6 Until now, there exist more than 460 compounds containing 9-AC ligand and its derivatives in the Cambridge Crystallographic Data Centre database. Hereinto, the photodimerization of the 9-AC ligand has probably been the most studied as it is prone to intermolecular [4+4] cycloaddition reactions after UV light irradiation, inducing mechanical bending, magnetic, and photoluminescent variations.7–11 Furthermore, the dimer analogue could also be reversibly returned to the monomer state via either a heat treatment or another light irradiation, making them potentially exploitable in dynamic optical crosslinking, molecular switches, energy storage, and mechanical devices.12–14 However, no one has reported the photochromic properties for 9-AC itself, even though the color changes have been naked-eye detected in some anthracene-based complexes after light irradiation from intermolecular [4+4] cycloaddition reactions.15
Photochromic materials, particularly the organic photochromic architectures, have continued to attract considerable attention owing to their potential applications in photonic materials and optical memory devices.16 Currently, numerous photochromic organic ligands, including diarylethenes, spiropyrans, azo compounds, anils, polycyclic quinones, viologens, triarylmethanes, and others have been discovered, accompanied with the mechanism involving either pericyclic reactions, cis–trans photoisomerization, or electron transfer processes.17–19 As for the electron transfer photochromic family, such as viologens, electron donors and acceptors are required to stabilize the photogenerated radicals and actuate the color variations.20,21 As an important aspect, the large π-conjugated ligand should be considered to construct electron transfer photochromic materials because they can provide a general and reliable strategy to obtain stable radicals. Therefore, the 9-AC molecule with large π-conjugated characterization may finish the task and realize the electron transfer photochromism. Via the photogenerated radicals, the photomagnetic phenomenon could be observed while introducing the paramagnetic metal ions into the 9-AC ligand-constructed complexes. Moreover, the photogenerated radicals and photocycloaddition may be simultaneously produced in the 9-AC components and control the magnetic couplings of metal centers, further promoting the performance of the photochromic and photomagnetic properties.
In this study, we demonstrate that 9-AC can show unexpected ambient photochromism in the crystalline state (Fig. S1, ESI†). The photoactive color variation originates from the intra- or/and intermolecular electron transfer process accompanied with photogenerated stable radicals, which was confirmed by time-dependent fluorescence, UV-Vis, IR, electron spin resonance (ESR) spectroscopy, magnetic measurements, and molecular orbital calculations. The variations of photochromism in a triclinic and monoclinic configuration were also explored, and the mixture of both forms exhibited more obvious photochromic property with a larger fluorescence intensity decrease after light irradiation. Furthermore, a mononuclear complex Ni(TEA)2·2(9-AC) (1; TEA = triethanolamine) constructed from 9-AC, TEA, and Ni(NO3)2·6H2O was designed and synthesized to confirm the photogenerated radicals, accompanied with the obvious photochromism and photomagnetism at ambient conditions.
The 9-AC ligand was purchased commercially and recrystallized in methanol with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. 1a).22 The configuration and phase purity were confirmed via powder X-ray diffraction analyses (Fig. S2, ESI†). After 250 W Xe-lamp light illumination, the yellow 9-AC samples, which were ground into powders, turned deeper to dark yellow, showing the characteristics of photoactivity (Fig. 1b and Fig. S3, ESI†). The solid-state UV-Vis spectra were first recorded to explore the light-induced color variations. As shown in Fig. 1c, a broad peak centered at about 500 nm appeared after photoactivation using a Xe-lamp, and this absorption peak tended to increase as the irradiation continued, suggesting the generation of 9-AC˙ radicals. The room temperature photoluminescence of the triclinic 9-AC molecule was in situ investigated at the polycrystalline state to gain more information about the photogenerated radicals (Fig. 1d). Fluorescence spectra showed that maximum emission at 496 nm occurred for this ligand upon excitation at 368 nm, which is ascribed to the intraligand π⋯π* or n⋯π* transitions. Upon Xe-lamp light irradiation, the fluorescence intensity gradually decreased by 13.1% of the initial value with prolonged irradiation time. This phenomenon should be due to the fluorescent 9-AC ligand changing to a non-fluorescent 9-AC˙ radical, which in turn quenched the fluorescence. To exclude the intermolecular [4+4] cycloaddition reactions of the 9-AC molecules, the solid-state IR spectra were recorded at ambient conditions. Since new PXRD peaks and transition bands could be obviously observed for the photochemical dimer,11,23 the same PXRD and IR spectra before and after illumination (Fig. S2 and S4, ESI†) ruled out the photodimerization and implied the formation of stable radicals.
space group (Fig. 1a).22 The configuration and phase purity were confirmed via powder X-ray diffraction analyses (Fig. S2, ESI†). After 250 W Xe-lamp light illumination, the yellow 9-AC samples, which were ground into powders, turned deeper to dark yellow, showing the characteristics of photoactivity (Fig. 1b and Fig. S3, ESI†). The solid-state UV-Vis spectra were first recorded to explore the light-induced color variations. As shown in Fig. 1c, a broad peak centered at about 500 nm appeared after photoactivation using a Xe-lamp, and this absorption peak tended to increase as the irradiation continued, suggesting the generation of 9-AC˙ radicals. The room temperature photoluminescence of the triclinic 9-AC molecule was in situ investigated at the polycrystalline state to gain more information about the photogenerated radicals (Fig. 1d). Fluorescence spectra showed that maximum emission at 496 nm occurred for this ligand upon excitation at 368 nm, which is ascribed to the intraligand π⋯π* or n⋯π* transitions. Upon Xe-lamp light irradiation, the fluorescence intensity gradually decreased by 13.1% of the initial value with prolonged irradiation time. This phenomenon should be due to the fluorescent 9-AC ligand changing to a non-fluorescent 9-AC˙ radical, which in turn quenched the fluorescence. To exclude the intermolecular [4+4] cycloaddition reactions of the 9-AC molecules, the solid-state IR spectra were recorded at ambient conditions. Since new PXRD peaks and transition bands could be obviously observed for the photochemical dimer,11,23 the same PXRD and IR spectra before and after illumination (Fig. S2 and S4, ESI†) ruled out the photodimerization and implied the formation of stable radicals.
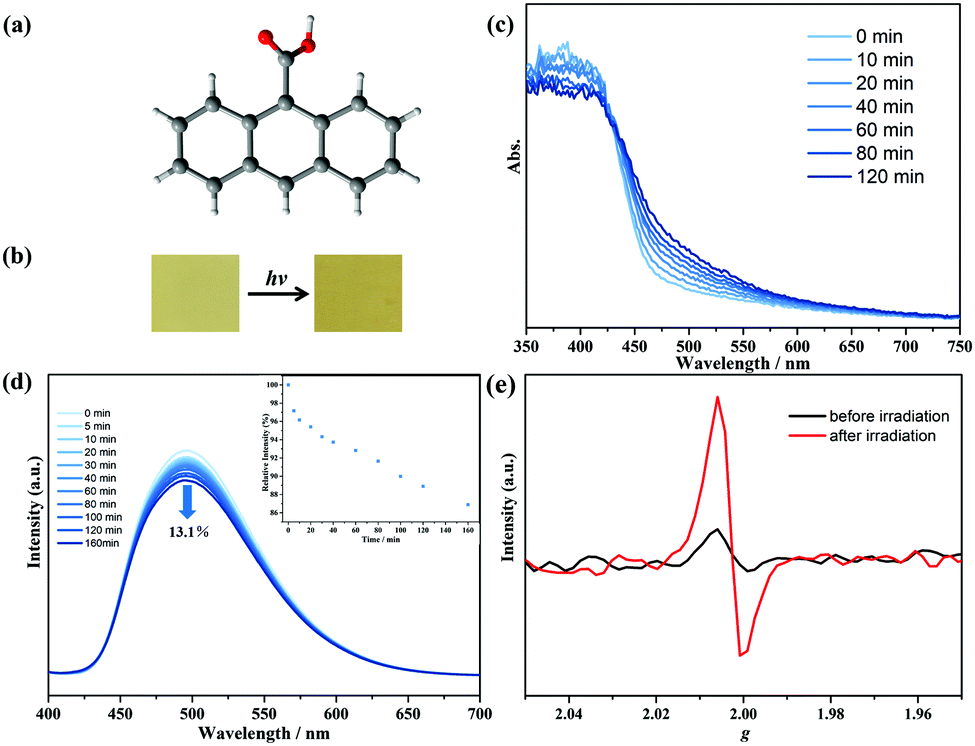 | ||
| Fig. 1 (a) The structure of the triclinic 9-AC molecule; (b) the photochromic properties of triclinic 9-AC; (c) time-dependent UV-Vis spectra of triclinic 9-AC at solid state upon light irradiation; (d) time-dependent fluorescent spectra of triclinic 9-AC at solid state upon light irradiation when excited at 368 nm. Inset: The variation of the maximum emission of triclinic 9-AC at different irradiation times; (e) the ESR spectra of triclinic 9-AC before and after light irradiation at solid state. | ||
The 9-AC˙ radicals were also confirmed by temperature-dependent susceptibility measurements using the polycrystalline samples. The as-prepared triclinic sample was diamagnetic. However, the χT value at 300 K was detected to be 0.08 cm3 K mol−1 after irradiation, suggesting the generation of 9-AC˙ radicals (Fig. S5, ESI†). To demonstrate the photogenerated radicals, solid state X-bond ESR curves of the polycrystalline triclinic 9-AC samples were measured at ambient conditions. As shown in Fig. 1e, the ESR signal was silent for the as-prepared samples, while a remarkable signal at g = 2.0025 appeared after Xe-lamp illumination, which is the characteristic of free radical species. For further verification of the electron transfer processes of the photoactive 9-AC molecule, the spatial distributions of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were calculated using the Gaussian 09 program and the basis set B3LYP/6-311G(d) method, indicating that the electron distribution of the HOMO was mainly located on the anthracene units, while the electron distribution of the carboxyl group appeared in LUMO (Fig. S6 and Table S1, ESI†). The typical localization of HOMO and LUMO suggests that the HOMO–LUMO transition becomes the intramolecular charge transfer transition.24,25 The calculated results suggested that electron transfer occurred from the anthracene component to the carboxyl group, finally forming the radical structure.
In general, an extremely conjugated structure can induce spin delocalization to a large π-system, preventing the generated active radicals from undergoing dimerization and polymerization reactions.26–29 For the 9-AC molecule, the photogenerated radical was relatively stable in the solid state under ambient conditions. The deeper color of 9-AC after irradiation maintained the charge separated state even after it was kept in dark for 5 weeks. Compared with most organic radicals having short lifetimes in air, the stability of the 9-AC˙ radical should be due to spin delocalization originating from the large π-conjugated anthracene components and strong interchain π–π interactions (Fig. S7, ESI†), similar to other reported stable organic and organometallic radical species.30–32
It is well known that both a triclinic and a monoclinic polymorphism can be concomitant in the 9-AC molecule under different solvent crystallization.22,33 To further check a possible difference of photochromism between these two forms, the 9-AC crystals with the monoclinic P21/n space group were synthesized from ethyl acetate.7 The yellow monoclinic crystals, confirmed by PXRD and IR measurements (Fig. 2a and Fig. S8, ESI†), became deeper after 2 h of Xe lamp light irradiation. However, the monoclinic one showed a decrease of about 15.3% in fluorescence intensity and needed less irradiation time to achieve the stable state (Fig. 2b). The anomaly should be due to the difference in the intermolecular interactions (Fig. S9 and S10, ESI†). The monoclinic structure adopted a nearer hydrogen bonding distance of 1.5476 (287) Å than that of the triclinic one (1.7259 (524) Å), providing a more efficient electron transfer pathway during the generation of radicals. The variation between the two configurations also suggested that the intermolecular electron transfer participates in the photochromic behavior of the 9-AC molecule. Furthermore, when the 9-AC ligand was recrystallized in the solvents of methanol and water (v/v = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), both the triclinic and monoclinic forms coexisted in this product, as confirmed by PXRD and light irradiated IR (Fig. 2a and Fig. S11, ESI†). Interestingly, the fluorescence intensity decreased by 25.6% with the duration of light irradiation (Fig. 2c), larger than the single triclinic and monoclinic phases. To further confirm this large decrease, the 1:1 mixture of triclinic and monoclinic forms, recrystallized from methanol and ethyl acetate, respectively, were also prepared and confirmed by the measured PXRD. The in situ light actuated fluorescence was performed at room temperature. As shown in Fig. 2d, the magnitude of the fluorescence intensity sharply decreased to 41.5% of the initial value, about three times decrease in intensity than the single triclinic and monoclinic phases. The highest photochromic efficiency in the mixed form should have originated from the nearer H-bonding distances, the stronger H-bonding interactions formed than in the separated triclinic/monoclinic ones, finally resulting in the much larger luminescence decrease after irradiation.
:1), both the triclinic and monoclinic forms coexisted in this product, as confirmed by PXRD and light irradiated IR (Fig. 2a and Fig. S11, ESI†). Interestingly, the fluorescence intensity decreased by 25.6% with the duration of light irradiation (Fig. 2c), larger than the single triclinic and monoclinic phases. To further confirm this large decrease, the 1:1 mixture of triclinic and monoclinic forms, recrystallized from methanol and ethyl acetate, respectively, were also prepared and confirmed by the measured PXRD. The in situ light actuated fluorescence was performed at room temperature. As shown in Fig. 2d, the magnitude of the fluorescence intensity sharply decreased to 41.5% of the initial value, about three times decrease in intensity than the single triclinic and monoclinic phases. The highest photochromic efficiency in the mixed form should have originated from the nearer H-bonding distances, the stronger H-bonding interactions formed than in the separated triclinic/monoclinic ones, finally resulting in the much larger luminescence decrease after irradiation.
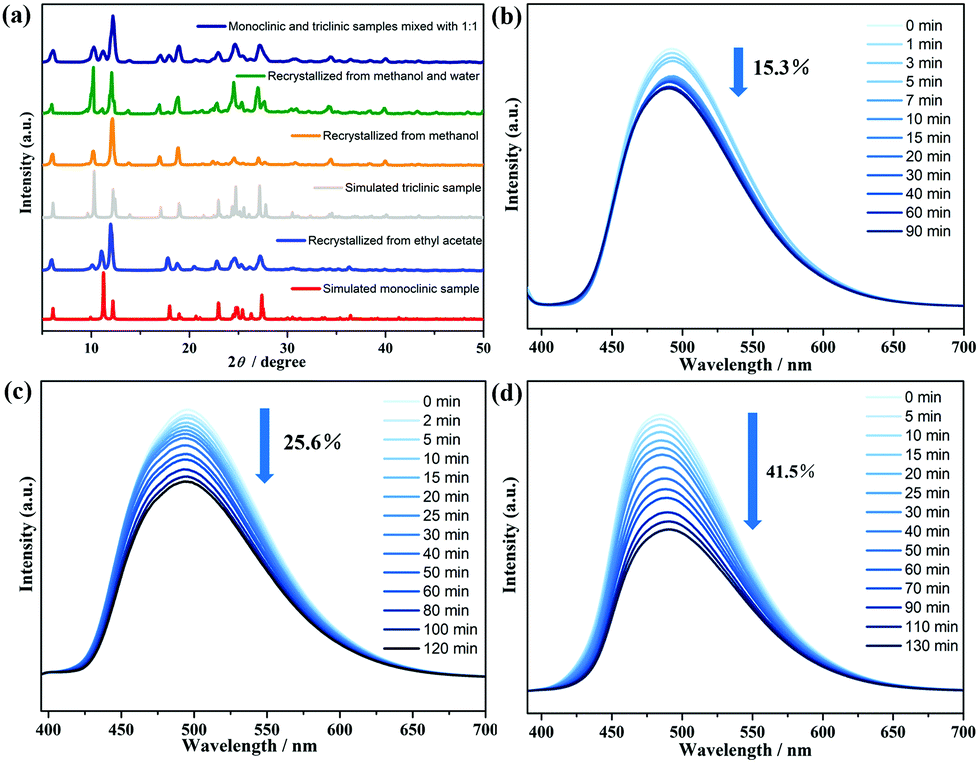 | ||
| Fig. 2 PXRD plots of different 9-AC samples (a); time-dependent fluorescent spectra upon light irradiation of monoclinic 9-AC (b), 9-AC samples recrystallized from methanol and water (c), monoclinic and triclinic samples mixed with 1:1 (d), respectively. | ||
Since the 9-AC molecule could produce stable radicals after light irradiation, the photochromic and photomagnetic behaviors may be realized via introducing this ligand into the paramagnetic ion-based complexes. Therefore, a mononuclear NiII-based complex 1 constructed by 9-AC, triethanolamine (TEA), and Ni(NO3)2·6H2O was obtained under hydrothermal reactions.
Single-crystal X-ray diffraction analysis demonstrated that compound 1 crystallized in the monoclinic P21/n space group. As shown in Fig. 3a, the structure consisted of one NiII center, two coordinated TEA molecules, and two-isolated 9-AC− groups. The Ni atom exhibited an [O4N2] octahedral geometry coordinated with two TEA ligands, and each TEA acted as a tridentate ligand through the binding N site and two hydroxyl O atoms. The Ni–O distances were in the range of 2.046(2)–2.0584(19) Å, with the Ni–N distance of 2.086(2) Å. It should be noted that the non-coordinated hydroxyl groups of the TEA ligands formed O1–H⋯O4 hydrogen bonds with the 9-AC− anions, while the coordinated hydroxyl groups also formed O2–H⋯O4 and O3–H⋯O5 hydrogen bonds with the 9-AC− anions (Fig. 3b and Table S4, ESI†). These strong intermolecular hydrogen bonds ranging from 2.594(3) to 2.679(3) Å linked the molecular units and provided an important way for the light actuated electron transfer process.
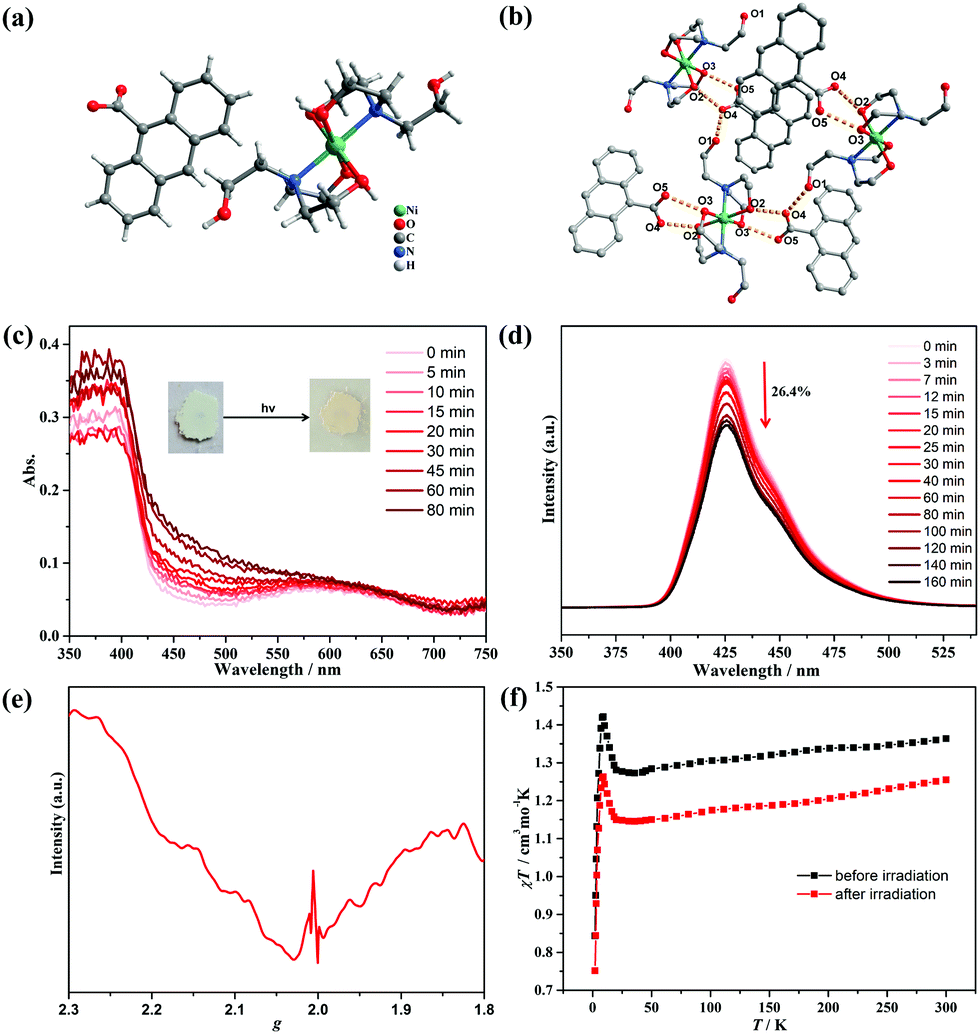 | ||
| Fig. 3 (a) Structure of compound 1; (b) H-bonding interactions of 1. H atoms are omitted for clarity; (c) time-dependent UV-Vis spectra of 1 at solid state upon light irradiation. Inset: The photochromic properties of 1; (d) time-dependent fluorescent spectra of 1 at solid state when excited at 280 nm; (e) the room temperature ESR spectra of compound 1 after light irradiation at a frequency of 9.84 GHz; (f) temperature-dependent susceptibilities of 1 under a dc magnetic field of 1000 Oe before and after light irradiation. | ||
The purity of compound 1 was firstly confirmed by PXRD measurements (Fig. S12, ESI†). Thermogravimetric analysis in the N2 atmosphere revealed that 1 was stable below 200 °C before decomposition (Fig. S13, ESI†). The photochromic behavior of compound 1 was then explored under ambient conditions. The powder samples exhibited color changes from light green to pale-yellow under irradiation by a 250 W Xe lamp within 20 min (Fig. S14, ESI†), indicating the light-induced electron transfer process. The time-dependent photochromic process was monitored by UV-vis absorption spectra (Fig. 3c). With the duration of light irradiation, the color of 1 changed to yellow, and the absorption peak centered at 460 nm became obviously stronger. This suggested the photogeneration of stable radicals. The same solid state PXRD and IR spectra before and after light irradiation precluded the intermolecular [4+4] cycloaddition reactions (Fig. S12 and S15, ESI†). The photoluminescence of compound 1 was also performed and exhibited high sensitivity towards UV light. As shown in Fig. 3d, compound 1 has fluorescence emission peaks at 426 nm with a shoulder at 450 nm. Moreover, the intensity of the main fluorescence peak gradually decreased to 26.4% of the initial value along with the irradiation time and confirmed the production of stable radicals. The ESR spectra of 1 were further measured after light irradiation to confirm the photogenerated radicals (Fig. 3e). The curve displayed a broad peak in the measured region, which should be ascribed to the NiII ions in the octahedral geometry. Surprisingly, a sharp peak at g = 2.004 appeared for the colored samples, demonstrating that the photochromic phenomenon was a light-induced electron transfer process, and the stable radicals were produced after Xe lamp illumination.
As a photogenerated spin carrier, the photomagnetic behavior was explored for 1 under a direct current magnetic field of 1000 Oe between 2–300 K (Fig. 3f). Before irradiation, the χT value was 1.35 cm3 K mol−1 at 300 K, slightly higher than the theoretical value of 1.21 cm3Kmol−1 expected for one high-spin NiII ion with S = 1 and g = 2.20.34 Upon cooling, the curve decreased to a minimum value of 1.27 cm3 K mol−1 at 35 K, and then continuously increased to 1.43 cm3 K mol−1 at 8.5 K, which is characteristic of ferrimagnetism between the NiII centers. Finally, the curve sharply decreased to 0.85 cm3 K mol−1 at 2 K, which originated from the zero-field splitting or intermolecular antiferromagnetic interactions.35 The magnetization values in the M–H curve at 2 K exhibited a quick increase to 1.79 Nβ at 50 kOe (Fig. S16, ESI†). For the irradiated samples, the χT–T and M–H curves exhibited a similar tendency with the one before irradiation. However, the χT values decreased to 1.25 cm3 K mol−1 at 300 K, and this obvious decrease should have stemmed from antiferromagnetic coupling interactions between the NiII ions and photogenerated radicals.
In summary, we discovered the crystalline 9-AC ligand as a novel room temperature electron transfer photoactive compound. This molecule changed to the radical analogue after Xe-lamp light irradiation at ambient conditions, which was demonstrated by PXRD analyses, UV-Vis, IR, fluorescence, ESR spectra, magnetic measurements, and molecular orbital calculations. A comparison of the triclinic and monoclinic crystalline configurations demonstrated that a higher photochromic efficiency appeared in the monoclinic form, which originated from the difference in the intermolecular interactions between the two configurations. Furthermore, a mononuclear complex based on the 9-AC ligand was synthesized with remarkable photochromic and photomagnetic behaviors, further demonstrating the light actuated radical property of the 9-AC molecule. For the first time, the 9-AC ligand and the constructed complexes were demonstrated to show radical actuated photochromic and photomagnetic behaviors, finding a new simple radical-actuated photochromic molecule for constructing a novel system of photochromic and photomagnetic materials. Further exploration of the photochromism of 9-AC based complexes is on the way.
This work was supported by the National Natural Science Foundation of China (21901133 and 22071126).
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. T. Burdzinski, R. Ramnauth, M. H. Chisholm and T. L. Gustafson, J. Am. Chem. Soc., 2006, 128, 6776 CrossRef CAS PubMed.
- A. A. Alaimo, D. Takahashi, L. Cunha-Silva, G. Christou and T. C. Stamatatos, Inorg. Chem., 2015, 54, 2137 CrossRef CAS PubMed.
- M. J. Byrnes, M. H. Chisholm, J. A. Gallucci, Y. Liu, R. Ramnauth and C. Turro, J. Am. Chem. Soc., 2005, 127, 17343 CrossRef CAS PubMed.
- C. X. Chen, Q. F. Qiu, M. Pan, C. C. Cao, N. X. Zhu, H. P. Wang, J. J. Jiang, Z. W. Wei and C. Y. Su, Chem. Commun., 2018, 54, 13666 RSC.
- T. Yamamoto, S. Yagyu and Y. Tezuka, J. Am. Chem. Soc., 2016, 138, 3904 CrossRef CAS PubMed.
- Y. Yang, X. Fang, S. S. Zhao, F. Y. Bai and D. Yan, Chem. Commun., 2020, 56, 5267 RSC.
- L. J. Fitzgerald and R. E. Gerkin, Acta Crystallogr., Sect. C: Struct. Chem., 1997, 53, 71 CrossRef PubMed.
- C. S. Liu, J. J. Wang, L. F. Yan, Z. Chang, X. H. Bu, E. C. Saundo and J. Ribas, Inorg. Chem., 2007, 46, 6299 CrossRef CAS PubMed.
- J. Wang, S. Zhang, S. Xu, A. Li, B. Li, L. Ye, Y. Geng, Y. Tian and W. Xu, Adv. Opt. Mater., 2020, 8, 1901280 CrossRef CAS.
- X. D. Huang, G. H. Wen, S. S. Bao, J. G. Jia and L. M. Zheng, Chem. Sci., 2021, 12, 929 RSC.
- X. D. Huang, Y. Xu, K. Fan, S. S. Bao, M. Kurmoo and L. M. Zheng, Angew. Chem., Int. Ed., 2018, 28, 8577 CrossRef PubMed.
- R. O. Al-Kaysi and C. J. Bardeen, Adv. Mater., 2010, 19, 1276 CrossRef.
- T. Salzillo, E. Venuti, C. Femoni, R. G. Della Valle, R. Tarroni and A. Brillante, Cryst. Growth Des., 2017, 17, 3361 CrossRef CAS.
- T. Salzillo and A. Brillante, CrystEngComm, 2019, 21, 3127 RSC.
- X. D. Huang, M. Kurmoo, S. S. Bao, K. Fan, Y. Xu, Z. Hu and L. M. Zheng, Chem. Commun., 2018, 54, 3278 RSC.
- J. C. Crano and R. J. Guglielmetti, Top. Appl. Chem., 1999, 1, 376 Search PubMed.
- E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579 CAS.
- R. Pardo, M. Zayat and D. Levy, Chem. Soc. Rev., 2011, 40, 672 RSC.
- A. J. Liu, F. Xu, S. D. Han, J. Pan and G. M. Wang, Cryst. Growth Des., 2020, 20, 7350 CrossRef CAS.
- Y. J. Ma, J. X. Hu, S. D. Han, J. Pan and G. M. Wang, J. Am. Chem. Soc., 2020, 142, 2682 CrossRef CAS PubMed.
- J. K. Sun, X. D. Yang, G. Y. Yang and J. Zhang, Coord. Chem. Rev., 2017, 378, 533 CrossRef.
- E. Heller and G. M. J. Schmidt, Isr. J. Chem., 1971, 9, 449 CrossRef CAS.
- R. More, G. Busse, J. Hallmann, C. Paulmann and S. Techert, J. Phys. Chem. C, 2010, 114, 4142 CrossRef CAS.
- X. H. Jin, J. Wang, J. K. Sun, H. X. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2011, 50, 1149 CrossRef CAS PubMed.
- J. Wang, S. L. Li and X. M. Zhang, ACS Appl. Mater. Inter., 2016, 8, 24862 CrossRef CAS PubMed.
- J. Cho, R. Sarangi, H. Y. Kang, J. Y. Lee and W. Nam, J. Am. Chem. Soc., 2010, 132, 3246 CrossRef PubMed.
- K. Kenichi and O. Atsuhiro, Angew. Chem., Int. Ed., 2019, 58, 8978 CrossRef PubMed.
- J. Wu, C. Tao, Y. Li, J. Li and J. Yu, Chem. Sci., 2015, 6, 2922 RSC.
- J. X. Hu, X. F. Jiang, Y. J. Ma, X. R. Liu, B. D. Ge, A. N. Wang, Q. Wei and G. M. Wang, Sci. China: Chem., 2021, 64, 432 CrossRef CAS.
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Adv. Mater., 2020, 32, 1908015 CrossRef CAS.
- N. N. Zhang, R. J. Sa, S. S. Sun, M. D. Li, M. S. Wang and G. C. Guo, J. Mater. Chem. C, 2019, 7, 3100 RSC.
- S. Tang, H. Ruan, R. Feng, Y. Zhao, G. Tan, L. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 18224 CrossRef CAS PubMed.
- R. Moré, M. Scholz, G. Busse, L. Busse, C. Paulmann, M. Tolkiehn and S. Techert, Phys. Chem. Chem. Phys., 2012, 14, 10187 RSC.
- X. Li, Z. Yu, X. Li and X. Guo, Chem. – Eur. J., 2015, 21, 16593 CrossRef CAS PubMed.
- P. F. Zhuang, Y. J. Zhang, H. Zheng, C. Q. Jiao, L. Zhao, J. L. Wang, C. He, C. Y. Duan and T. Liu, Dalton Trans., 2015, 44, 3393 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2063584. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc00920f |
| This journal is © The Royal Society of Chemistry 2021 |