
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrids of cationic [4]helicene and N-heterocyclic carbene as ligands for complexes exhibiting (chir)optical properties in the far red spectral window†
Robert
Tarrieu
a,
Irene Hernandez
Delgado
b,
Francesco
Zinna
 bc,
Vincent
Dorcet
d,
Sophie
Colombel-Rouen
a,
Christophe
Crévisy
*a,
Olivier
Baslé
*ae,
Johann
Bosson
*b and
Jérôme
Lacour
*b
bc,
Vincent
Dorcet
d,
Sophie
Colombel-Rouen
a,
Christophe
Crévisy
*a,
Olivier
Baslé
*ae,
Johann
Bosson
*b and
Jérôme
Lacour
*b
aUniv Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS, ISCR - UMR 6226, Rennes F-35000, France. E-mail: christophe.crevisy@ensc-rennes.fr
bDepartment of Organic Chemistry, University of Geneva, Quai Ernest Ansermet, 30, Geneva 4 CH-1211, Switzerland. E-mail: johann.bosson@unige.ch; jerome.lacour@unige.ch
cDipartimento di Chimica e Chimica Industriale, University of Pisa, via G. Moruzzi 13, Pisa, Italy
dUniv Rennes, CNRS, ISCR - UMR 6226, Rennes F-35000, France
eLCC-CNRS, Université de Toulouse, CNRS, UPS, Toulouse, France. E-mail: olivier.basle@lcc-toulouse.fr
First published on 11th March 2021
Abstract
Synthesis, electronic and structural properties of a chiral NHC bearing a N-bonded cationic [4]helicene moiety are reported. This ligand is used to construct AuI, AuIII and RhI complexes exhibiting far-red (chir)optical properties regardless of the metal.
In recent decades, N-heterocyclic carbenes (NHCs) have become ubiquitous ligands able, in the context of optical devices, to form robust organometallic complexes with luminescent properties.1–6 In most cases, the luminescence arises from the metal center and its coordination sphere, and it occurs predominantly at high energy (UV or blue-green spectral range). In the meantime, new NHC structures affording unique stereo-electronic properties have been prepared,7,8 and unsymmetrical unsaturated N-heterocyclic carbenes (U2-NHCs) in particular,9–13 allowing for instance the introduction of cycloalkyl or cationic cyclopropenium chains onto the diaminocarbene unit (e.g., A and B, Fig. 1).14 In addition, chiral NHCs are readily prepared,15–18 some of them using helicenes as stereogenic elements.19–24 Helicenes are ortho-condensed polyaromatic molecules, inherently chiral due to the steric repulsion of their skeleton termini.25–31 Generally, they display absorption and emission properties in the UV and blue regions of light,32–34 with redshifted (chir)optical properties for some particular derivatives.35–39 In previous helicene-NHC based organometallic complexes, effective metal centered (chir)optical properties were engineered and happened at high energy.19,20,23,24 Herein, a different approach is detailed based on ligand-controlled luminescence. Synthetically, using a one-pot multicomponent procedure,13 cationic [4]helicene aniline 1-NH2(BF4)38,40 and cyclododecylamine are combined to generate imidazolium 2·H(PF6)2 (Fig. 1). The hybrid NHC ligand 2(PF6) is readily formed and affords stable RhI, AuI and AuIII complexes. The NHC steric and electronic properties are discussed. Low energy absorption and emission of the cationic [4]helicene moiety are maintained. Regardless of the organometallic species, intense (chir)optical properties are observed in the far-red spectral region, which can be beneficial for bio-related applications.41
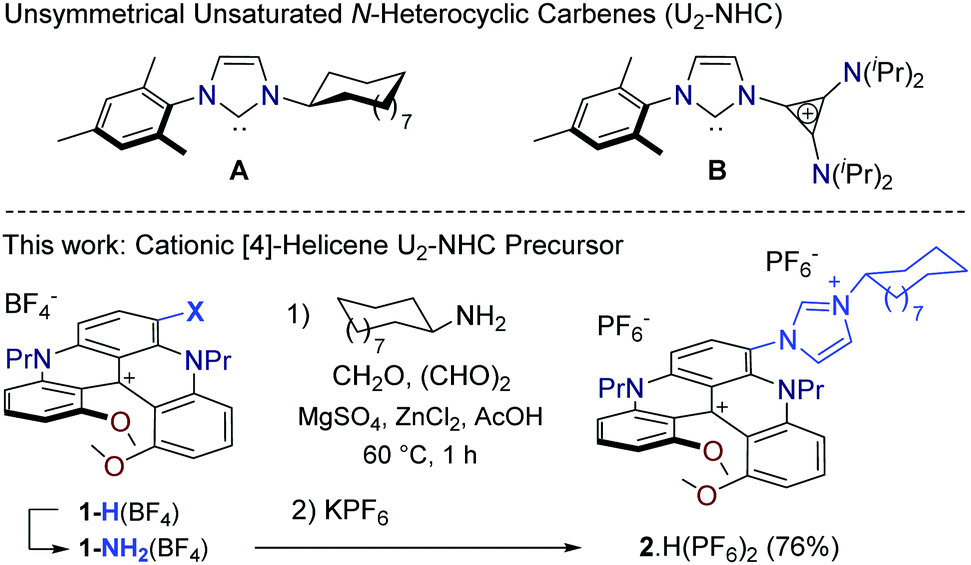 | ||
| Fig. 1 Top: Examples of unsymmetrical unsaturated N-heterocyclic carbenes (U2-NHCs). Bottom: Preparation of bis cationic imidazolium salt 2·H(PF6)2 from cationic [4]helicene aniline 1-NH2(BF4), (P)-configuration shown arbitrarily. | ||
For the preparation of targeted imidazolium salt 2·H(PF6)2, unfunctionalized cationic diaza[4]helicene 1-H(BF4) (Fig. 1, X = H),40 was converted into aniline 1-NH2(BF4) using a reported two-step sequence (nitration-reduction).38 Then, using conditions particularly efficient for the preparation of U2-NHCs precursors made of sterically hindered anilines,131-NH2(BF4) was engaged with equimolar amounts of cyclododecylamine, glyoxal and formaldehyde in the presence of zinc chloride and acetic acid. After counterion metathesis, the unsymmetrical bis cationic imidazolium salt 2·H(PF6)2 was obtained with a high selectivity (93%) over the bis cyclododecyl counterpart and was isolated as a dark blue-green solid in 76% yield (Fig. 1). As for NHC B (Fig. 1),14 the presence of the positive charge on the [4]helicene moiety was not a problem for the construction of organometallic complexes. After the in situ generation of free NHC 2(PF6) using potassium tert-butoxide as a base, standard metalation procedures afforded selenium adduct 3(PF6), rhodium(I) 4(PF6), gold(I) 5(PF6) and gold(III) 6(PF6)2 complexes in good yields (51–81%, Fig. 2); all compounds display a dynamic atropisomerism that will be detailed in the following paragraphs. In practice, trapping of 2(PF6) with selenium (excess) led to selenourea 3(PF6) as a mixture of two inseparable stereoisomers (75%, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, 1H and 77Se NMR, Fig. S26 and S30, ESI†). Reaction of 2(PF6) with [Rh(cod)Cl]2 dimer yielded (NHC)RhCl(cod) complex 4(PF6) (69% yield; atropisomeric ratio a.r. 3:2). The preparation of gold complexes 5(PF6) and 6(PF6)2 was achieved by reactions with AuCl·SMe2 and bis-cyclometalated 2,6-diphenylpyridyl gold(III) complex, respectively. 5(PF6) was isolated in 51% yield (a.r. 3:2), whereas 6(PF6)2 was formed in 81% yield, only one apparent stereoisomer being observed in this case at 298 K (vide infra).
:2 ratio, 1H and 77Se NMR, Fig. S26 and S30, ESI†). Reaction of 2(PF6) with [Rh(cod)Cl]2 dimer yielded (NHC)RhCl(cod) complex 4(PF6) (69% yield; atropisomeric ratio a.r. 3:2). The preparation of gold complexes 5(PF6) and 6(PF6)2 was achieved by reactions with AuCl·SMe2 and bis-cyclometalated 2,6-diphenylpyridyl gold(III) complex, respectively. 5(PF6) was isolated in 51% yield (a.r. 3:2), whereas 6(PF6)2 was formed in 81% yield, only one apparent stereoisomer being observed in this case at 298 K (vide infra).
 | ||
| Fig. 2 Selenium adduct 3(PF6), rhodium(I) 4(PF6), gold(I) and gold(III) complexes 5(PF6) and 6(PF6)2 prepared from 2·H(PF6)2. (P) enantiomers shown. a.r.: atropisomeric ratio. | ||
As above-noted, compounds 3 to 5 displayed in NMR spectroscopy two sets of signals on the NMR time scale. This behavior results from the restricted rotation around the C–N bond42 linking the helicene and the imidazole moieties. In fact, the steric hindrance about the C–N bond generates pairs of atropisomers, for instance of (aS,P) and (aR,P) configurations (Fig. 3); the atropisomeric ratio depending upon the encumbrance of the substituted carbene fragment. To explain the situation, care must be taken to consider NMR solution and X-ray crystallographic studies that have previously indicated that substituents at position of 6 of the helicene induce a strong conformational constraint onto the neighboring N-alkyl chains.38 The impacted propyl residues display gauche conformations (instead of classical anti conformations) and, consequently, they fold above the helical core (Fig. 3, blue propyl chain). This is particularly well observed in 1H NMR spectroscopy with the terminal methyl groups being shifted at lower frequency by −0.8 ppm. As a consequence, strong steric interactions occur between the curved propyl chain and metal coordination spheres at immediate proximity favoring the less hindered conformations that orientate the propyl chain and metal centers in opposite directions. Considering a (P) configuration for the helicene, the (aS,P) conformation is then favored over the diastereomeric (aR,P) geometry (Fig. 3, right vs. left).
 | ||
| Fig. 3 Atropisomers (aS,P) and (aR,P) formed with helicenes of (P)-configuration. | ||
The atropisomeric situation was investigated in silico. Conformational search (B3LYP/6-311G**) for a simplified version of selenium adduct (P)-3 was performed (see ESI†). The two conformers depicted in Fig. 3 were found to be the most stable structures, the aS isomer being more stable by 0.2 kcal mol−1, which corresponds to an equilibrium ratio of 0.58:0.42 at 25 °C. This is in good agreement with the experimental 0.6:0.4 value (vide supra). Moreover, the transition state of the aS → aR isomerization was located at +17.2 kcal mol−1 which is consistent with a facile epimerization at room temperature and yet with the possibility to distinguish the two species on the NMR time scale.43 Similarly, for 4 and 5, the calculated ratios were found to be in good agreement with 1H NMR spectroscopy (Table S1, ESI†). For compound 6, to our initial surprise, the computed energy difference between the stereoisomers was only of 0.1 kcal mol−1 (a.r. 0.54:0.46 at 298 K). Such a ratio could only be confirmed experimentally at lower temperature (a.r. 0.52:0.48 at 233 K, see Fig. S43, ESI†).44 Solid state structures of 3(PF6) and complex 4(PF6) were further obtained by X-ray diffraction analysis (Fig. 2, see also ESI,† for topographic steric maps and associated %Vbur values).45 In both cases, a single atropisomer was found in the crystal packing with a configuration opposite to that predicted computationally. It is most probably the result of intermolecular packing interactions and it also confirms the facile epimerization.
NHC ligands are reputed to stabilize organometallic complexes via the formation of strong metal carbene bonds that are related to the σ-donor character of the carbenic position and its ability to accept π-back-bonding from the metal center. Considering the unusual cationic character of NHC 2(PF6), the electronic properties of such species were investigated and are compiled in Table 1. First, the σ-donor ability of the NHC was assessed by measuring the 1JCH coupling constant between the carbon and hydrogen atom at the pre-carbenic position of precursor 2·H(PF6)2.46,47 The value of 1JCH = 224 Hz measured for 2·H(PF6)2 was found slightly superior to that of A (1JCH = 222 Hz) bearing the neutral mesityl fragment, and inferior to the 229 Hz reported for the cyclopropylium moiety B,14 highlighting the somewhat low withdrawing character of cationic [4]helicene substituent. The π-character of NHCs can be tabulated using 77Se NMR spectroscopy.48,49 The two isomers of the NHC-Se adduct 3(PF6) exhibited 77Se resonances at 65 and 49 ppm, downfield shifted compared to A-Se adduct (δSe = 10 ppm) revealing a slight increase of the π-character as compared to this neutral species, but less pronounced than for cationic B-Se adduct (122 ppm).14 Finally, the Tolman Electronic Parameters (TEP) value of NHC 2(PF6) was also investigated. The TEP allows to describe the effects of the combination of the σ-donor and π-acceptor characters modulated by the sterics of the ligand.50 For that purpose, the (NHC)RhCl(cod)complex 4(PF6) was converted under a carbon monoxide atmosphere to the corresponding (NHC)RhCl(CO)2 complex and allowed in situ measurement of the average IR stretching of the carbonyl ligands (Fig. S19, ESI†). The calculated TEP value of 2055.1 cm−1 between neutral unsaturated NHCs (i.e. TEP 2050.5 cm−1 for A) and cationic NHC B (2057 cm−1) is consistent with measured 1JCH and 77Se values indicating that the positive charge has a significant effect on the electronic properties of 2, lowering thus the σ-donation and increasing the π-acceptation, effect however less pronounced than that observed in the cationic B.
The optical properties of compounds 2·H(PF6)2 to 6(PF6)2 were recorded in acetonitrile solutions and compared to that of parent cationic[4]helicene 1-H(BF4) (Fig. 4, top and Table 2). The functionalization of 1-H (λabs: 616 nm, λem: 667 nm) with electron-withdrawing groups (EWG) at position 6 is documented to induce rather strong hypsochromic shifts in absorption and emission.382·H(PF6)2 exhibits a moderately intense absorption band in the orange-red region (λabs: 599 nm, ε: 11500 M cm−1) and fluoresces in the far red (ϕflu: 0.26 at 644 nm). The slight blue shifts observed as compared to 1-H indicate that the imidazolium cationic part can be described as a mild EWG, comparable in intensity to a ketone moiety.38 The lowest energy transition of the selenium adduct 3(PF6) is centered on 613 nm (ε = 9300 M cm−1) but this compound is essentially non-emissive.51 As expected, the characteristic optical properties of the diaza[4]helicene 1-H are maintained in the organometallic complexes 4–6 with only a marginal influence of the metallic centers (Table 2). The lowest energy transitions are centered between 600 (6(PF6)2) and 609 (4(PF6)) nm with ε values spanning from 9800 to 13200 M−1 cm−1. Similarly, no significant changes are observed in emission, all derivatives being efficient fluorophores in the far red (ϕflu: 0.14–0.23, λem 644–653 nm).
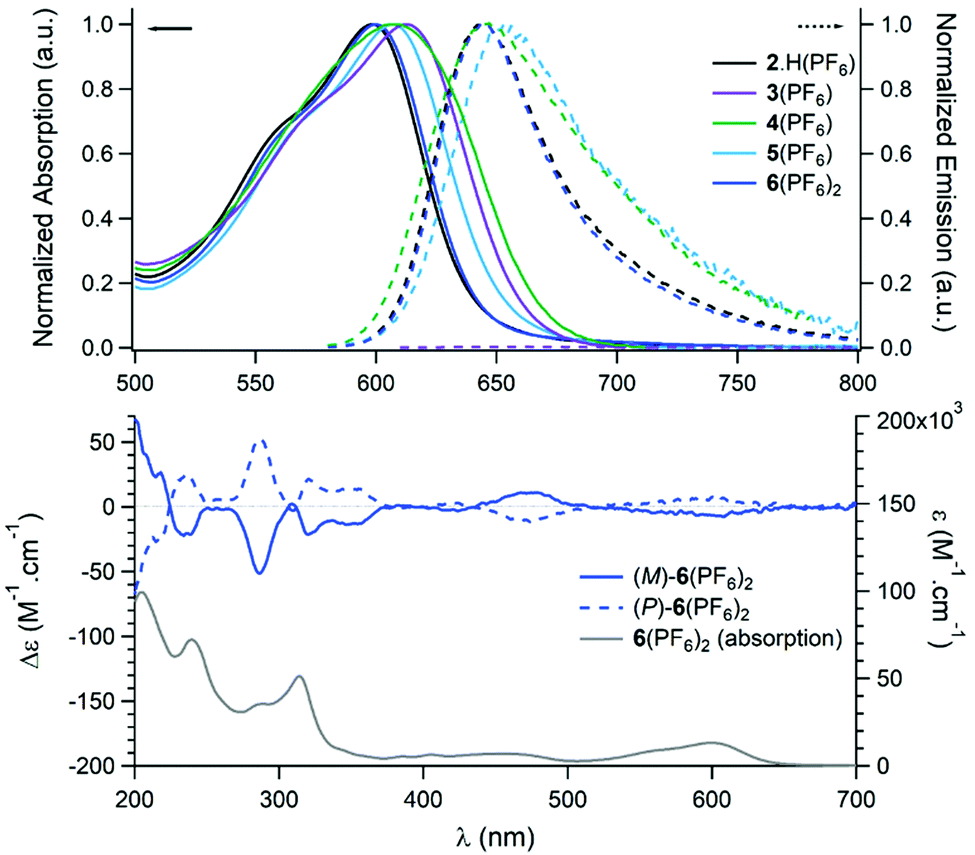 | ||
| Fig. 4 Top: Normalized absorption (plain lines) and emission (dashed lines) spectra of 2·H(PF6) – 6(PF6)2 recorded in acetonitrile (C ca. 10−5 M). Bottom: ECD spectra of (M)-6(PF6)2 and (P)-6(PF6)2 (blue lines) and absorption spectrum of 6(PF6)2 (grey line), acetonitrile, (C ca. 10−5 M). | ||
| λ abs (nm) | ε (M cm−1) | λ em (nm) | ϕ flu | |
|---|---|---|---|---|
| a Quantum yields versus cresyl violet (ϕflu 0.54 in MeOH). | ||||
| 1-H(BF4) | 616 | 14000 |
667 | 0.13 |
| 2·H(PF6)2 | 599 | 11500 |
644 | 0.26 |
| 3(PF6) | 613 | 9300 | — | — |
| 4(PF6) | 609 | 13000 |
646 | 0.14 |
| 5(PF6) | 607 | 9800 | 653 | 0.15 |
| 6(PF6)2 | 600 | 13200 |
644 | 0.23 |
AuIII complex 6(PF6)2 was selected to investigate the chiroptical properties of this family of compounds (Fig. 4, bottom). The two enriched (M) and (P) enantiomers of 6(PF6)2 were prepared from the corresponding (M)- and (P)-1-H. As usual for cationic helicenes,37,38,52,53 the two enantiomers of 6(PF6)2 exhibit strong specific rotations,  = −4000 and +4300 for the (M) and (P) enantiomers, respectively. In Electronic Circular Dichroism (ECD) and in the visible range, 6(PF6)2 exhibits two rather strong Cotton effects centered at 600 nm (|Δε| = 8 M−1 cm−1) and at 479 nm (|Δε| = 11 M−1 cm−1). In the UV region, Cotton effects typical of helicene skeletons are observed at 237, 287 and 320 nm (|Δε| = 23, 52 and 21 M−1 cm−1, respectively). The Circularly Polarized Luminescence (CPL) of 6(PF6)2 was also measured in acetonitrile solution (Fig. S9, ESI†). This species displays a glum value of ±2 × 10−4, which is the typical magnitude for such [4]helicenes. The CPL signal is centered on the maximum emission wavelength, which is strongly red-shifted as compared to most carbohelicenes.54
= −4000 and +4300 for the (M) and (P) enantiomers, respectively. In Electronic Circular Dichroism (ECD) and in the visible range, 6(PF6)2 exhibits two rather strong Cotton effects centered at 600 nm (|Δε| = 8 M−1 cm−1) and at 479 nm (|Δε| = 11 M−1 cm−1). In the UV region, Cotton effects typical of helicene skeletons are observed at 237, 287 and 320 nm (|Δε| = 23, 52 and 21 M−1 cm−1, respectively). The Circularly Polarized Luminescence (CPL) of 6(PF6)2 was also measured in acetonitrile solution (Fig. S9, ESI†). This species displays a glum value of ±2 × 10−4, which is the typical magnitude for such [4]helicenes. The CPL signal is centered on the maximum emission wavelength, which is strongly red-shifted as compared to most carbohelicenes.54
In summary, a dicationic helicene-imidazolium hybrid was prepared following a high yielding multicomponent synthetic procedure, revealing a ligand characterized by an overall richness comparable to common neutral unsaturated NHCs and a certain structural flexibility. This helicene-NHC was used for the construction of RhI, AuI and AuIII complexes that exhibit ligand controlled chiroptical properties in the far-red window.
This work was supported by the CNRS, the Ecole Nationale Supérieure de Chimie de Rennes, the Région Bretagne and Demeta (ARED No. COH14007 “NHC-MET” grant to R.T.), the University of Geneva and the Swiss National Science Foundation (grants 172497 and 184843).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- T. Zou, F. F. Hung, C. Yang and C. M. Che, Luminescent and Photoactive Transition Metal Complexes as Biomolecular Probes and Cellular Reagents, 2015, vol. 165, pp. 181–203 Search PubMed.
- C. Hemmert and H. Gornitzka, Dalton Trans., 2016, 45, 440–447 RSC.
- C. Hille and F. E. Kuhn, Dalton Trans., 2016, 45, 15–31 RSC.
- P. V. Simpson, M. Falasca and M. Massi, Chem. Commun., 2018, 54, 12429–12438 RSC.
- I. Omae, Coord. Chem. Rev., 2016, 310, 154–169 CrossRef CAS.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- A. Fürstner, M. Alcarazo, V. César and C. W. Lehmann, Chem. Commun., 2006, 2176–2178 RSC.
- P. Queval, C. Jahier, M. Rouen, I. Artur, J.-C. Legeay, L. Falivene, L. Toupet, C. Crévisy, L. Cavallo, O. Baslé and M. Mauduit, Angew. Chem., Int. Ed., 2013, 52, 14103–14107 CrossRef CAS PubMed.
- S. Li, F. Yang, T. Lv, J. Lan, G. Gao and J. You, Chem. Commun., 2014, 50, 3941–3943 RSC.
- C. Jahier-Diallo, M. S. T. Morin, P. Queval, M. Rouen, I. Artur, P. Querard, L. Toupet, C. Crévisy, O. Baslé and M. Mauduit, Chem. – Eur. J., 2015, 21, 993–997 CrossRef CAS PubMed.
- R. Tarrieu, A. Dumas, J. Thongpaen, T. Vives, T. Roisnel, V. Dorcet, C. Crévisy, O. Baslé and M. Mauduit, J. Org. Chem., 2017, 82, 1880–1887 CrossRef CAS PubMed.
- C. Barthes, C. Duhayon, Y. Canac and V. César, Chem. Commun., 2020, 56, 3305–3308 RSC.
- F. Wang, L.-j. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804–853 CrossRef CAS.
- D. Janssen-Müller, C. Schlepphorst and F. Glorius, Chem. Soc. Rev., 2017, 46, 4845–4854 RSC.
- P. J. Czerwiński and M. Michalak, Synthesis, 2019, 1689–1714 Search PubMed.
- J. Thongpaen, R. Manguin and O. Baslé, Angew. Chem., Int. Ed., 2020, 59, 10242–10251 CrossRef CAS PubMed.
- N. Hellou, C. Jahier-Diallo, O. Baslé, M. Srebro-Hooper, L. Toupet, T. Roisnel, E. Caytan, C. Roussel, N. Vanthuyne, J. Autschbach, M. Mauduit and J. Crassous, Chem. Commun., 2016, 52, 9243–9246 RSC.
- N. Hellou, M. Srebro-Hooper, L. Favereau, F. Zinna, E. Caytan, L. Toupet, V. Dorcet, M. Jean, N. Vanthuyne, J. A. G. Williams, L. Di Bari, J. Autschbach and J. Crassous, Angew. Chem., Int. Ed., 2017, 56, 8236–8239 CrossRef CAS PubMed.
- I. G. Sanchez, M. Samal, J. Nejedly, M. Karras, J. Klivar, J. Rybacek, M. Budesinsky, L. Bednarova, B. Seidlerova, I. G. Stara and I. Stary, Chem. Commun., 2017, 53, 4370–4373 RSC.
- M. Karras, M. Dabrowski, R. Pohl, J. Rybacek, J. Vacek, L. Bednarova, K. Grela, I. Stary, I. G. Stara and B. Schmidt, Chem. – Eur. J., 2018, 24, 10994–10998 CrossRef CAS PubMed.
- N. Hafedh, L. Favereau, E. Caytan, T. Roisnel, M. Jean, N. Vanthuyne, F. Aloui and J. Crassous, Chirality, 2019, 31, 1005–1013 CrossRef CAS PubMed.
- E. S. Gauthier, L. Abella, N. Hellou, B. Darquie, E. Caytan, T. Roisnel, N. Vanthuyne, L. Favereau, M. Srebro-Hooper, J. A. G. Williams, J. Autschbach and J. Crassous, Angew. Chem., Int. Ed., 2020, 59, 8394–8400 CrossRef CAS PubMed.
- Y. Shen and C. F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- P. Aillard, A. Voituriez and A. Marinetti, Dalton Trans., 2014, 43, 15263–15278 RSC.
- N. Saleh, C. S. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC.
- J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824–2840 RSC.
- J. OuYang and J. Crassous, Coord. Chem. Rev., 2018, 376, 533–547 CrossRef CAS.
- I. G. Stara and I. Stary, Acc. Chem. Res., 2020, 53, 144–158 CrossRef CAS PubMed.
- E. M. Sanchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. – Eur. J., 2015, 21, 13488–13500 CrossRef CAS PubMed.
- H. Isla and J. Crassous, C. R. Chim., 2016, 19, 39–49 CrossRef CAS.
- W. L. Zhao, M. Li, H. Y. Lu and C. F. Chen, Chem. Commun., 2019, 55, 13793–13803 RSC.
- M. Li, Y. Niu, X. Zhu, Q. Peng, H. Y. Lu, A. Xia and C. F. Chen, Chem. Commun., 2014, 50, 2993–2995 RSC.
- M. Li, W. Yao, J. D. Chen, H. Y. Lu, Y. S. Zhao and C. F. Chen, J. Mater. Chem. C, 2014, 2, 8373–8380 RSC.
- J. Bosson, G. M. Labrador, S. Pascal, F. A. Miannay, O. Yushchenko, H. Li, L. Bouffier, N. Sojic, R. C. Tovar, G. Muller, D. Jacquemin, A. D. Laurent, B. Le Guennic, E. Vauthey and J. Lacour, Chem. – Eur. J., 2016, 22, 18394–18403 CrossRef CAS PubMed.
- I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guenee, C. Nancoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC.
- M. Li, H. Y. Lu, C. Zhang, L. Shi, Z. Tang and C. F. Chen, Chem. Commun., 2016, 52, 9921–9924 RSC.
- C. Herse, D. Bas, F. C. Krebs, T. Burgi, J. Weber, T. Wesolowski, B. W. Laursen and J. Lacour, Angew. Chem., Int. Ed., 2003, 42, 3162–3166 CrossRef CAS.
- S. Wang, B. Li and F. Zhang, ACS Cent. Sci., 2020, 6, 1302–1316 CrossRef CAS.
- L. Kong, J. Morvan, D. Pichon, M. Jean, M. Albalat, T. Vives, S. Colombel-Rouen, M. Giorgi, V. Dorcet, T. Roisnel, C. Crévisy, D. Nuel, P. Nava, S. Humbel, N. Vanthuyne, M. Mauduit and H. Clavier, J. Am. Chem. Soc., 2020, 142, 93–98 CrossRef CAS PubMed.
- Considering first-order kinetics, kinetic constant of 1.50 s−1 and a half-life t1/2 of 0.46 s at 298 K are calculated.
- In addition, the activation energy is quite lower (+13.5 kcal mol−1) due possibly to steric buttressing.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- G. Meng, L. Kakalis, S. P. Nolan and M. Szostak, Tetrahedron Lett., 2019, 60, 378–381 CrossRef CAS.
- K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2416–2425 CrossRef CAS.
- S. V. C. Vummaleti, D. J. Nelson, A. Poater, A. Gomez-Suarez, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and L. Cavallo, Chem. Sci., 2015, 6, 1895–1904 RSC.
- A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- (a) Heavy atom effect ie, G. C. Hoover and D. S. Seferos, Chem. Sci., 2019, 10, 9182–9188 RSC; (b) or photoinduced electron transfer ie. M. Rosenberg, A. K. R. Junker, T. J. Sørensen and B. W. Laursen, ChemPhotoChem, 2019, 3, 233–242 CrossRef CAS; (c) K. Koren, N. K. G. Salinas, M. Santella, M. Mosshammer, M.-C. Mueller, R. I. Dmitriev, S. M. Borisov, M. Kühl and B. W. Laursen, Dyes Pigm., 2020, 173, 107866 CrossRef CAS can be invoked for the non-emissive character.
- S. Pascal, C. Besnard, F. Zinna, L. Di Bari, B. Le Guennic, D. Jacquemin and J. Lacour, Org. Biomol. Chem., 2016, 14, 4590–4594 RSC.
- G. M. Labrador, J. Bosson, Z. S. Breitbach, Y. Lim, E. R. Francotte, R. Sabia, C. Villani, D. W. Armstrong and J. Lacour, Chirality, 2016, 28, 282–289 CrossRef CAS PubMed.
- C.-F. Chen and Y. Shen, Helicene Chemistry, Springer, Berlin, Heidelberg, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental conditions, full characterizations of new compounds; UV-Vis, ECD, fluorescence and CPL spectra; computational details. CCDC 2061397 and 2061398. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc00898f |
| This journal is © The Royal Society of Chemistry 2021 |