
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermally-induced hyperbranching of bromine-containing polyesters by insertion of in situ generated chain-end carbenes†
Panagiotis
Bexis
ab,
Maria C.
Arno
 bc,
Craig A.
Bell
ade,
Anthony W.
Thomas
a and
Andrew P.
Dove
*b
bc,
Craig A.
Bell
ade,
Anthony W.
Thomas
a and
Andrew P.
Dove
*b
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: A.Dove@bham.ac.uk
cInstitute of Cancer and Genomic Science, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
dCentre for Advanced Imaging, The University of Queensland, Brisbane, QLD 4072, Australia
eAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia
First published on 24th March 2021
Abstract
Hyperbranched, biodegradable PCL-based polymers are obtained through a random but invasive migration of an in situ generated carbene end group which is unmasked via the thermolysis of its precursor diazirine moiety. These hyperbranched cores are used as macroinitiators for ‘grafting-from’ polymerisation using controlled radical polymerisation to achieve amphiphilic copolymers which can subsequently be self-assembled into spherical core–shell micelles.
Highly branched 3D macromolecular structures, including dendrimers and hyperbranched polymers (HBPs) have emerged as an important class of materials1,2 on account of the large number of terminal functional groups, globular three-dimensional structures, and low intrinsic viscosities that they display compared to linear polymer species.3,4 Of these species, HBPs exhibit the advantages of low (or no) chain entanglements, as observed in linear polymers, as well as low melt and solution viscosity, high solubility, and a wide variety of terminal functional groups which serve as handles for further modification.5–7 Furthermore, in contrast to perfectly branched and monodisperse dendrimers consisting of only dendritic units and terminal units, HBPs are composed of dendritic units, linear units and terminal units, and display a randomly branched structure with a lower degree of branching (DB). More importantly, compared to the tedious and complicated synthetic procedures of dendrimers, the synthesis of HBPs is often based on simple one-pot reactions, requiring minimal or no further purification.2,8–10
Very recently, new progress in the self-assembly11–13 of HBPs has been achieved. HBPs have proven to be excellent precursors in supramolecular self-assembly, allowing the generation of a diverse range of supramolecular structures and hybrids at all scales and dimensions. As such, assemblies obtained from HBPs have recently found applications in the biomedical field, owing to their ease of synthesis, their large number of end groups which provides a platform for the conjugation of functional moieties, their controlled responsive nature and their ability to incorporate a multiple array of guest molecules through covalent or noncovalent approaches.5,9,14–16
Diazirines17 are a class of functional groups characterised by three-membered heterocycles featuring an azo group and an sp3 hybridised carbon atom.18,19 The defining property of diazirines is their ability to form highly reactive carbenes with concomitant release of molecular nitrogen upon suitable activation via exposure to light, high temperature20 or ultrasonication. Carbenes are capable of reacting (via insertion) with saturated hydrocarbons and by addition with unsaturated hydrocarbons, including aromatic systems.21 This ease of insertion makes diazirines the perfect tool for photoaffinity labelling,22,23 crosslinking,24 surface modification of polymers25,26 and polymer branching when incorporated within functional monomers.27,28
Herein, we describe for the first time the use of a hydroxyl-functional aromatic diazirine as an initiator for the organocatalytic ring-opening polymerisation (ROP) of a bromine-functional caprolactone monomer, α-bromo-ε-caprolactone (BrCL) alongside copolymerisation with ε-caprolactone (CL). The diazirine moieties undergo thermolysis to generate a highly reactive carbene species which can directly attack the long macromolecular chains, forming branching points both inter- and intra-molecularly. The hyperbranched bromine-bearing hydrophobic polyesters are subsequently utilised as macroinitiators to graft hydrophilic PEG branches, yielding amphiphilic materials able to self-assemble into spherical core–shell micelles.29
The BrCL monomer,30,31 selected as the alkyl bromide, is a handle that enables copper-mediated radical polymerisations used in grafting techniques,32–35 and is largely unreactive towards carbene insertion;23,26–28,45,50 the polycaprolactone (PCL)-based polymer serves as the hydrophobic and biodegradable segment in the material.30,36–39 Organocatalytic ROP40 was selected for the initial polymerisation of the cyclic ester monomers. The ROP of CL and the subsequent copolymerisation of CL and BrCL was attempted using diphenyl phosphate (DPP) as the catalyst41–43 and 4-[3-(trifluoromethyl)-3H-diazirin-3-yl] benzyl alcohol (TFDBA) as the initiator (Scheme S1, ESI† and Fig. 1A).
 | ||
| Fig. 1 (A) Copolymerisation of CL and BrCL catalysed by DPP and initiated by TFDBA leading to the gradient blocky α-diazirine copolymers; (B) thermal activation of the diazirine leading to the unmasking of the carbene and the subsequent hyperbranching of the linear precursor polymers; (C) application of Cu(0)-RDRP techniques enables ‘grafting-from’ the PCL-based materials, leading to amphiphilic polymers. | ||
CL was first polymerised targeting degrees of polymerisation (DPs) of 5, 10 and 20 (Table S1, ESI†) to assess the viability of using an alcohol-functional diazirine as an initiator for ROP. The observed low dispersity and monomodal molar mass distribution of the polymers served as an indication of well-controlled polymerisations (Fig. S1, ESI†), and 1H and 13C NMR spectroscopy provided structural data as well as the experimental molar mass of the polymers. The 19F NMR spectrum showed a single resonance at δ = −65 ppm in all synthesised polymers, which proved the specific incorporation of the CF3 group of the diazirine only in the desired α-position of the polymer chain (Fig. S2–S8, ESI†).
Hyperbranching of the TFDBA-initiated PCLs was initially investigated (Fig. 1B) by heating the polymers in bulk at 100 °C for 6 h. The conversion of the diazirine functionality was qualitatively monitored by 19F NMR spectroscopy for the PCL10 (Fig. S9 and S10, ESI†) by monitoring the disappearance of the CF3 group on the initiator. New peaks appeared between δ = −79 and −66 ppm throughout the thermolysis, indicating that a change in fluorine environment was occurring – in line with carbenes being generated in situ and inserting into the polymer backbone causing the branching (Scheme S2, ESI†), further corroborated by 13C NMR spectroscopic analysis (Fig. S6 and S11, ESI†).44 At the reaction end-point (Fig. S10, ESI†), the parent fluorine peak had almost disappeared for all PCL polymers, indicating near quantitative loss of the starting diazirine functionality. As a consequence of the random character of this hyperbranching methodology,27,28 and given the structural diversity of the linear and branched units within the polyester architecture, the 1H and 13C NMR spectra of the branched PCL homopolymers provided little structural information regarding the extent of branching (Fig. S11–S18, ESI†).45,46 Clear trends of increasing molar mass (Mn, Mw) and dispersity (ĐM)47,48 were observed for all PCLs as the reaction proceeded, as evidenced by triple detection size exclusion chromatography (SEC-td) (Fig. S19, S20 and Table S2, ESI†). This is attributed to the formation of random branches within the polymer scaffold triggered by carbene-initiated C–H and C–O bond insertions.27,28,49 SEC-td monitoring of the PCL10 polymer revealed a gradual molar mass and dispersity increase (Fig. S21 and Table S3, ESI†) in close agreement with the conversion of the diazirine functionality, as shown from the 19F NMR spectra. Analysis of the Mark–Houwink50 plot of the hyperbranched PCL materials revealed that despite an almost ten-fold increase in Mw compared to their linear precursors, the HB polymers retained similar values of η (intrinsic viscosity) (Fig. S22, ESI†). From these analyses, the alpha (α) values of these polymers were calculated to be between 0.38 and 0.48 (Tables S2 and S3, ESI†), which is a clear indication of branching.51 Analysis of a linear PCL control that contained a benzyl α-end group (no diazirine) and an experimental Mw close to that of the HBPs (Mw = 11.4 kDa, ĐM = 1.03) revealed α = 0.78 (Fig. S23A, ESI†). After heating this polymer in bulk at 100 °C for 6 h, SEC-td showed a significant reduction in the molar mass of the polymer. At the same time, the dispersity increased, but the Mark–Houwink alpha value retained its high value (α = 0.73). Thus, without the diazirine end group, the polymer remains linear and undergoes extensive transesterification but no hyperbranching, which only occurs in the presence of the diazirine moiety (Fig. S23B, ESI†).
In order to create gradient copolymers of BrCL and CL suitable to create core–shell structures, ROP and hyperbranching was conducted in a similar manner. Targeting a total degree of polymerisation (DP) of 10, three different feed ratios of monomer (CL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BrCL) were used, 25:75, 50:50, 75:25 (Table S4, ESI†). An increased catalyst loading ([TFDBA]0:[DPP]0 = 1:2) was used in order to accelerate the reactions due to the lower propagation rate of BrCL.30 After 4 h, the conversion of CL was near quantitative in all three polymerisations, whereas the conversion of BrCL was ca. 60% in all three experiments. After quenching the polymerisations and purification of the polymers, 1H, 13C and 19F NMR spectroscopy was used to show retention of diazirine functionality on the α-chain end of the copolymers as well as to characterise the copolymers (Fig. S24–S32, ESI†). Analysis by SEC-td revealed the experimental Mn of the copolymers to be in close accordance with the calculated values (between 1.8–2.9 kDa depending on the desired composition), with dispersities slightly higher than expected (ĐM = 1.37–1.59), possibly as a consequence of the extended reaction times causing unavoidable transesterification side-reactions and a gradient-block copolymer microstructure (Fig. S33, ESI†).30
:BrCL) were used, 25:75, 50:50, 75:25 (Table S4, ESI†). An increased catalyst loading ([TFDBA]0:[DPP]0 = 1:2) was used in order to accelerate the reactions due to the lower propagation rate of BrCL.30 After 4 h, the conversion of CL was near quantitative in all three polymerisations, whereas the conversion of BrCL was ca. 60% in all three experiments. After quenching the polymerisations and purification of the polymers, 1H, 13C and 19F NMR spectroscopy was used to show retention of diazirine functionality on the α-chain end of the copolymers as well as to characterise the copolymers (Fig. S24–S32, ESI†). Analysis by SEC-td revealed the experimental Mn of the copolymers to be in close accordance with the calculated values (between 1.8–2.9 kDa depending on the desired composition), with dispersities slightly higher than expected (ĐM = 1.37–1.59), possibly as a consequence of the extended reaction times causing unavoidable transesterification side-reactions and a gradient-block copolymer microstructure (Fig. S33, ESI†).30
The hyperbranching of the P(CL-co-BrCL) copolymers followed by 19F NMR spectroscopy showing the unmasking of the carbene and its gradual insertion into the saturated polyester backbone until almost full completion (Fig. 2 and Fig. S36, S41, ESI†). The 1H and 13C NMR spectra of the resulting hyperbranched polymers (Fig. S34, S35, S37–S40, ESI†) do not elucidate the degree of branching but do confirm the retention of the C–Br resonance at δ = 45.9 ppm. Significant increases in molar masses and dispersities compared to their linear precursors (SEC-td) further confirms the formation and subsequent reaction of the carbene to form branched structures. (Fig. S42–S44 and Table S5, ESI†). Mark–Houwink analysis showed that the intrinsic viscosity of the HBPs was maintained at similar values compared to their linear copolymer precursors, while their larger molar mass and the obtained low α values (< 0.5) confirmed their hyperbranched nature (Fig. S46 and Table S6, ESI†). Further analysis of the available SEC and 1H NMR spectroscopy data enabled the elucidation of the average number of arms (Narms)52–54 of each of the HBPs (Table S6, ESI†).47 Based on the absolute molar masses (obtained via SEC-td), dispersity (ĐM,RI obtained by refractive index detection55) of the HBPs, and the Mn of the linear precursor polymers (obtained by NMR spectroscopic analysis), Narms was found to decrease from 12 to 7, as the content of BrCL decreased for each polymer. This phenomenon can be ascribed to the bromine's steric hindrance, which prevents the approach of carbenes to free C–C or C–H bonds, thus limiting the branching events.
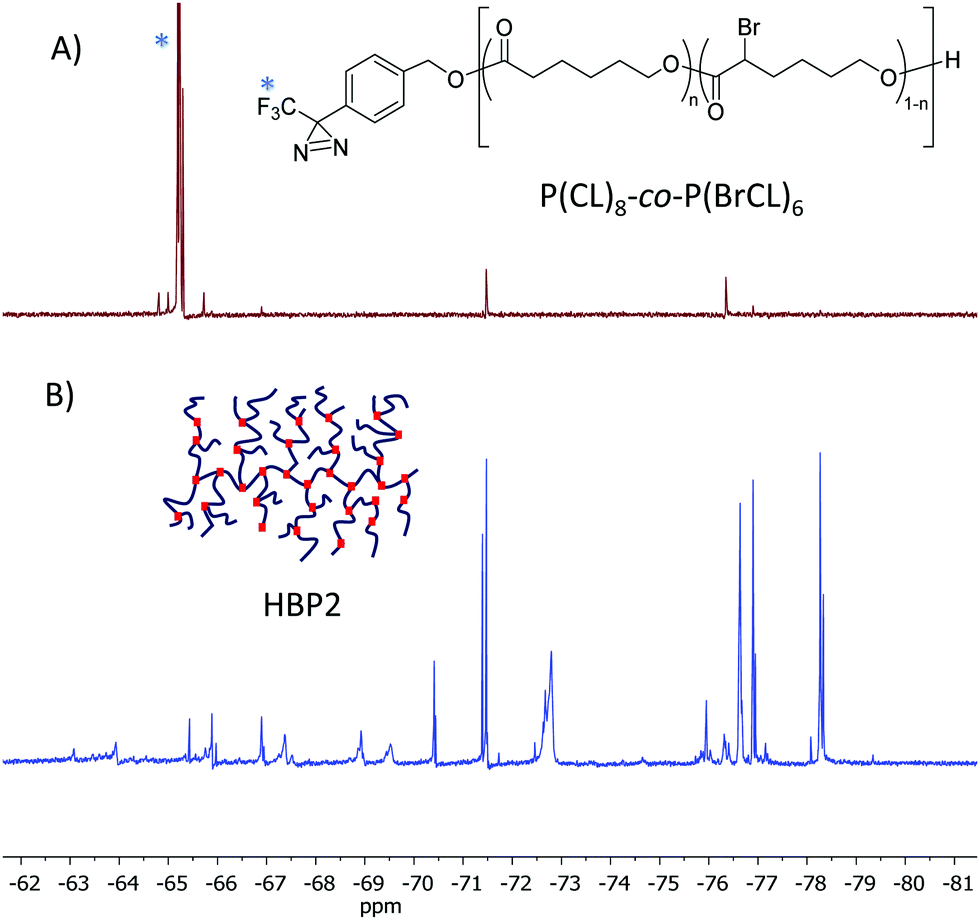 | ||
| Fig. 2 Stacked 19F NMR spectra of the linear P(CL)8-co-P(BrCL)6 (top) and the branched structure HBP2 (bottom, after 6 h of branching – end point data). The diazirine has decomposed, thereby unmasking the carbene, which has then inserted in various C–H and C–O bonds of the polymer, thus creating a random hyperbranched structure (CDCl3). | ||
The HB polymers bearing pendent Br (Fig. S45, ESI†) can subsequently serve as suitable macroinitiators for the application of Cu(0)-mediated RDRP,56–61 thus enabling ‘grafting-from’ the PCL-based polymers.30 Judicious choice of the synthetic method, grafting density, composition and length of the polymer backbone and side-arms, allows graft copolymers with unique structural characteristics and a range of functionalities to be prepared.62–65 HBP2 was selected as the macroinitiator, and methyl acrylate (MA) was first polymerised in a grafting-from approach, targeting 20 MA grafted units per initiating site (Br unit) for each arm. After 24 h the monomer conversion was >99%, and upon purification the polymer appeared as a waxy transparent solid. 1H NMR spectroscopy confirmed the synthesis of PMA (Fig. S47, ESI†), and successful grafting was proved by SEC-td as the molecular weight distribution of the starting polymer completely shifted to higher molecular weight, while its dispersity decreased significantly (ĐM,SEC-td = 1.5, Fig. S51A, ESI†).
Following the successful grafting of MA, poly(ethylene glycol) methyl ether acrylate (PEGMEA) was polymerised targeting 10, 20 and 50 units per initiating site for each arm. All reactions reached quantitative conversion after 24 h. 1H NMR spectroscopy and SEC-td of the purified materials again proved the successful grafting of the acrylate had occurred (Fig. S51B–D, S48–S50 and Table S7, ESI†). The alpha (α) values of all PEGMEA-grafted polymers were maintained below 0.5, thus confirming the retention of their branched structure. The multimodality of the SEC chromatograms is possibly an effect of the random spatial nature of hyperbranching causing the irregular placement of initiating sites for the grafting of brushes, which in turn might cause intense radical coupling, termination events or extremely high grafting density.
Self-assembly was performed on the HBP2-g-PEGMEA50 amphiphilic polymer using a traditional solvent switch method by dissolving the polymer in THF and slowly adding water to induce micelle formation (Fig. 3A). Micelles of ∼50 nm in diameter were observed by dynamic light scattering (DLS) analysis, with spherical morphology confirmed by transmission electron microscopy (TEM) (Fig. 3B, C and Fig. S52, ESI†).
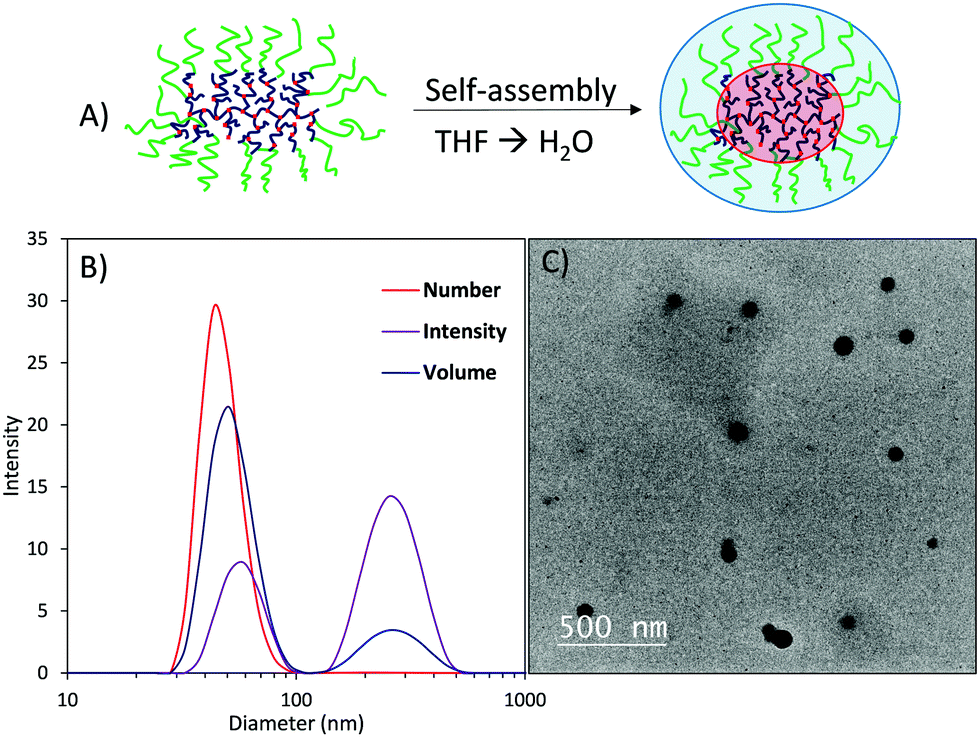 | ||
| Fig. 3 (A) Self-assembly of HBP2 using a solvent switch method (slow addition of H2O into THF/polymer solution) leading to the formation of spherical core–shell particles; (B) DLS analysis of HBP2-g-PEGMEA50 micelles (1 mg mL−1), and (C) TEM micrograph of HBP2-g-PEGMEA50 micelles. | ||
In conclusion, this work reports a new thermally induced methodology for preparing hyperbranched PCL-based materials. These bromine-bearing structures are shown to serve as ideal macroinitiators for the application of Cu(0)-RDRP techniques, which lead to further grafted polymers. These amphiphilic polymers of extremely high molar mass can be self-assembled into spherical core–shell particles, thus making them attractive candidates for further downstream bio-applications.
CAB acknowledges funding for this research from the National Health and Medical Research Council (APP1054569).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- M. Jikei and M. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233–1285 CrossRef CAS.
- A. B. Cook, R. Barbey, J. A. Burns and S. Perrier, Macromolecules, 2016, 49, 1296–1304 CrossRef CAS.
- B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2505–2525 CrossRef CAS.
- D. Wang, T. Zhao, X. Zhu, D. Yan and W. Wang, Chem. Soc. Rev., 2015, 44, 4023–4071 RSC.
- C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183–275 CrossRef CAS.
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257–1281 CrossRef CAS.
- D. Konkolewicz, M. J. Monteiro and S. Perrier, Macromolecules, 2011, 44, 7067–7087 CrossRef CAS.
- S. Peleshanko and V. V. Tsukruk, Prog. Polym. Sci., 2008, 33, 523–580 CrossRef CAS.
- Y. Zhou and D. Yan, Chem. Commun., 2009, 1172–1188, 10.1039/b814560c.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS PubMed.
- A. B. Cook and S. Perrier, Adv. Funct. Mater., 2019, 30, 1901001 CrossRef.
- Y. Zhou, W. Huang, J. Liu, X. Zhu and D. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS PubMed.
- B. I. Voit and A. Lederer, Chem. Rev., 2009, 109, 5924–5973 CrossRef CAS PubMed.
- A. Blencowe and W. Hayes, Soft Matter, 2005, 1, 178–205 RSC.
- S. R. Paulsen, Angew. Chem., Int. Ed. Engl., 1960, 72, 781 CrossRef CAS.
- E. Schmitz, Adv. Heterocycl. Chem., 1963, 2, 83–130 CrossRef CAS.
- M. T. H. Liu, M. Tencer and I. D. R. Stevens, J. Chem. Soc., Perkin Trans. 2, 1986, 211–214 RSC.
- R. A. Moss, Acc. Chem. Res., 1989, 22, 15–21 CrossRef CAS.
- H. Nakashima, M. Hashimoto, Y. Sadakane, T. Tomohiro and Y. Hatanaka, J. Am. Chem. Soc., 2006, 128, 15092–15093 CrossRef CAS PubMed.
- S.-S. Ge, B. Chen, Y.-Y. Wu, Q.-S. Long, Y.-L. Zhao, P.-Y. Wang and S. Yang, RSC Adv., 2018, 8, 29428–29454 RSC.
- D. P. Smith, J. Anderson, J. Plante, A. E. Ashcroft, S. E. Radford, A. J. Wilson and M. J. Parker, Chem. Commun., 2008, 5728–5730, 10.1039/B813504E.
- A. Blencowe, K. Cosstick and W. Hayes, New J. Chem., 2006, 30, 53–58 RSC.
- A. Blencowe, C. Blencowe, K. Cosstick and W. Hayes, React. Funct. Polym., 2008, 68, 868–875 CrossRef CAS.
- A. Blencowe, N. Caiulo, K. Cosstick, W. Fagour, P. Heath and W. Hayes, Macromolecules, 2007, 40, 939–949 CrossRef CAS.
- A. Blencowe, W. Fagour, C. Blencowe, K. Cosstick and W. Hayes, Org. Biomol. Chem., 2008, 6, 2327–2333 RSC.
- R. A. Ramli, RSC Adv., 2017, 7, 52632–52650 RSC.
- P. Bexis, A. W. Thomas, C. A. Bell and A. P. Dove, Polym. Chem., 2016, 7, 7126–7134 RSC.
- G. Wang, Y. Shi, Z. Fu, W. Yang, Q. Huang and Y. Zhang, Polymer, 2005, 46, 10601–10606 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3931–3939 CrossRef CAS.
- Q. Zhang, A. Anastasaki, G.-Z. Li, A. J. Haddleton, P. Wilson and D. M. Haddleton, Polym. Chem., 2014, 5, 3876–3883 RSC.
- E. Turan and T. Caykara, React. Funct. Polym., 2011, 71, 1089–1095 CrossRef CAS.
- G. Lligadas, S. Grama and V. Percec, Biomacromolecules, 2017, 18, 1039–1063 CrossRef CAS PubMed.
- N. López-Rodríguez, A. López-Arraiza, E. Meaurio and J. R. Sarasua, Polym. Eng. Sci., 2006, 46, 1299–1308 CrossRef.
- A. Höglund, M. Hakkarainen and A. C. Albertsson, J. Macromol. Sci., Part A: Pure Appl. Chem, 2007, 44, 1041–1046 CrossRef.
- H. Seyednejad, A. H. Ghassemi, C. F. van Nostrum, T. Vermonden and W. E. Hennink, J. Controlled Release, 2011, 152, 168–176 CrossRef CAS PubMed.
- S. Ramakrishna, J. Mayer, E. Wintermantel and K. W. Leong, Compos. Sci. Technol., 2001, 61, 1189–1224 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- F. Ercole, A. E. Rodda, L. Meagher, J. S. Forsythe and A. P. Dove, Polym. Chem., 2014, 5, 2809–2815 RSC.
- D. Delcroix, A. Couffin, N. Susperregui, C. Navarro, L. Maron, B. Martin-Vaca and D. Bourissou, Polym. Chem., 2011, 2, 2249–2256 RSC.
- K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999–2005 CrossRef CAS.
- H. Ismaili, S. Lee and M. S. Workentin, Langmuir, 2010, 26, 14958–14964 CrossRef CAS PubMed.
- C. J. Hawker, J. M. J. Frechet, R. B. Grubbs and J. Dao, J. Am. Chem. Soc., 1995, 117, 10763–10764 CrossRef CAS.
- J. M. J. Fréchet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080–1083 CrossRef PubMed.
- S. Podzimek and T. Vlcek, J. Appl. Polym. Sci., 2001, 82, 454–460 CrossRef CAS.
- S. Podzimek, T. Vlcek and C. Johann, J. Appl. Polym. Sci., 2001, 81, 1588–1594 CrossRef CAS.
- S. Ghiassian, H. Ismaili, B. D. W. Lubbock, J. W. Dube, P. J. Ragogna and M. S. Workentin, Langmuir, 2012, 28, 12326–12333 CrossRef CAS PubMed.
- A. C. Makan, T. Otte and H. Pasch, Macromolecules, 2012, 45, 5247–5259 CrossRef CAS.
- F. Xiang, T. Loontjens, E. Geladé and J. Vorenkamp, Macromol. Chem. Phys., 2012, 213, 1841–1850 CrossRef CAS.
- Y. Zheng, W. Turner, M. Zong, D. J. Irvine, S. M. Howdle and K. J. Thurecht, Macromolecules, 2011, 44, 1347–1354 CrossRef CAS.
- J. H. Tan, N. A. J. McMillan, E. Payne, C. Alexander, F. Heath, A. K. Whittaker and K. J. Thurecht, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2585–2595 CrossRef CAS.
- N. T. Nguyen, K. J. Thurecht, S. M. Howdle and D. J. Irvine, Polym. Chem., 2014, 5, 2997–3008 RSC.
- M. Gaborieau and P. Castignolles, Anal. Bioanal. Chem., 2011, 399, 1413–1423 CrossRef CAS PubMed.
- M. J. Monteiro, T. Guliashvili and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1835–1847 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen, G. Lligadas and V. Percec, Macromolecules, 2009, 42, 2379–2386 CrossRef CAS.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef CAS.
- N. H. Nguyen, M. E. Levere, J. Kulis, M. J. Monteiro and V. Percec, Macromolecules, 2012, 45, 4606–4622 CrossRef CAS.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- C. Feng, Y. Li, D. Yang, J. Hu, X. Zhang and X. Huang, Chem. Soc. Rev., 2011, 40, 1282–1295 RSC.
- H.-i. Lee, J. Pietrasik, S. S. Sheiko and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 24–44 CrossRef CAS.
- S. S. Sheiko, B. S. Sumerlin and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 759–785 CrossRef CAS.
- M. Zhang and A. H. E. Müller, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3461–3481 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc00821h |
| This journal is © The Royal Society of Chemistry 2021 |