
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the origin of photo-induced enhancement of oxidation catalysis at Mo(VI) centres of Ru(II)–Mo(VI) dyads†
Maryam
Nazari Haghighi Pashaki
 a,
Tae-Kyu
Choi
b,
Egmont J.
Rohwer
a,
Thomas
Feurer
a,
Anne-Kathrin
Duhme-Klair
*c,
Wojciech
Gawelda
*def and
Andrea
Cannizzo
*a
a,
Tae-Kyu
Choi
b,
Egmont J.
Rohwer
a,
Thomas
Feurer
a,
Anne-Kathrin
Duhme-Klair
*c,
Wojciech
Gawelda
*def and
Andrea
Cannizzo
*a
aInstitute of Applied Physics, University of Bern, Sidlerstrasse 5, CH-3012, Bern, Switzerland. E-mail: andrea.cannizzo@iap.unibe.ch
bEuropean XFEL, Holzkoppel 4, 22869 Schenefeld, Germany
cDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: anne.duhme-klair@york.ac.uk
dDepartamento de Química, Facultad de Ciencias, Universidad Autónoma de Madrid, Campus Cantoblanco, 28049 Madrid, Spain. E-mail: wojciech.gawelda@uam.es
eInstituto Madrileño de Estudios Avanzados en Nanociencia (IMDEA-Nanociencia), Campus Cantoblanco, 28049 Madrid, Spain
fFaculty of Physics, Adam Mickiewicz University, ul. Uniwersytetu Poznańskiego 2, 61-614 Poznan, Poland
First published on 23rd March 2021
Abstract
Photo-induced oxidation-enhancement in biomimetic bridged Ru(II)–Mo(VI) photo-catalyst is unexpectedly photo-activated in ps timescales. One-photon absorption generates an excited state where both photo-oxidized and photo-reduced catalytic centres are activated simultaneously and independently.
Metalloenzymes often regenerate their active sites via efficient electron transfer (eT) between spatially separated eT units.1 Thus, the comprehension of electronic relays between these components in bioinspired redox catalysts is attracting increasing interest.2,3 Electrochemical approaches to mimic this process include for instance the attachment of reversible eT components to metalloenzyme mimics.4–6
Photo-induced eT processes have also been explored to harness solar energy and to accelerate redox catalysis. For example, ruthenium-based photosensitizers linked to manganese-based photosystem II models were used to mimic the four-electron oxidation of two water molecules by light-induced eT.7 Similarly, hydrogenase mimics with attached reversible photosensitizers have been investigated with the aim of catalysing the light-driven production of H2.8,9 Photoredox processes10–12 have also been used to catalyse oxygenation reactions with environmentally benign O-atom sources, in particular 3O2 or H2O.13 Enemark and Kirk et al. demonstrated that oxo-molybdenum(V) can be photoactivated via an antenna-mediated eT process by covalently linking the oxo–Mo(V) unit to porphyrin-Fe(III) or Zn(II) complexes.14,15
Recently, Duhme-Klair et al. developed biomimetic molybdenum complexes16 with appended ruthenium-based photoactive units to facilitate oxygen atom transfer (OAT) catalysis via photo-induced eT (Fig. 1). They found that the attachment of a bis(bipyridyl)–phenanthroline ruthenium complex to a cis-dioxo Mo(VI) thiosemicarbazone allows the OAT from dimethyl sulfoxide to triphenyl phosphine (PPh3) to be accelerated upon irradiation with visible light. To explain their findings, specifically the photo-induced oxidation-enhancing (PIOE) effect,17 they proposed the scheme illustrated in Fig. 1: the Ru(II) is first photo-oxidized and one 2,2′-bipyridine(bpy) ligand is photo-reduced (1); the latter is then oxidized by an oxidizing agent (e.g. methyl viologen) (2), leading to the production of a highly reactive, one-electron oxidized catalytic Mo(VI) unit via an intramolecular eT toward the Ru(III) (3). Eventually a reducing agent, as for instance PPh3, is oxidized via the OAT from the activated cis-dioxo molybdenum unit (4).
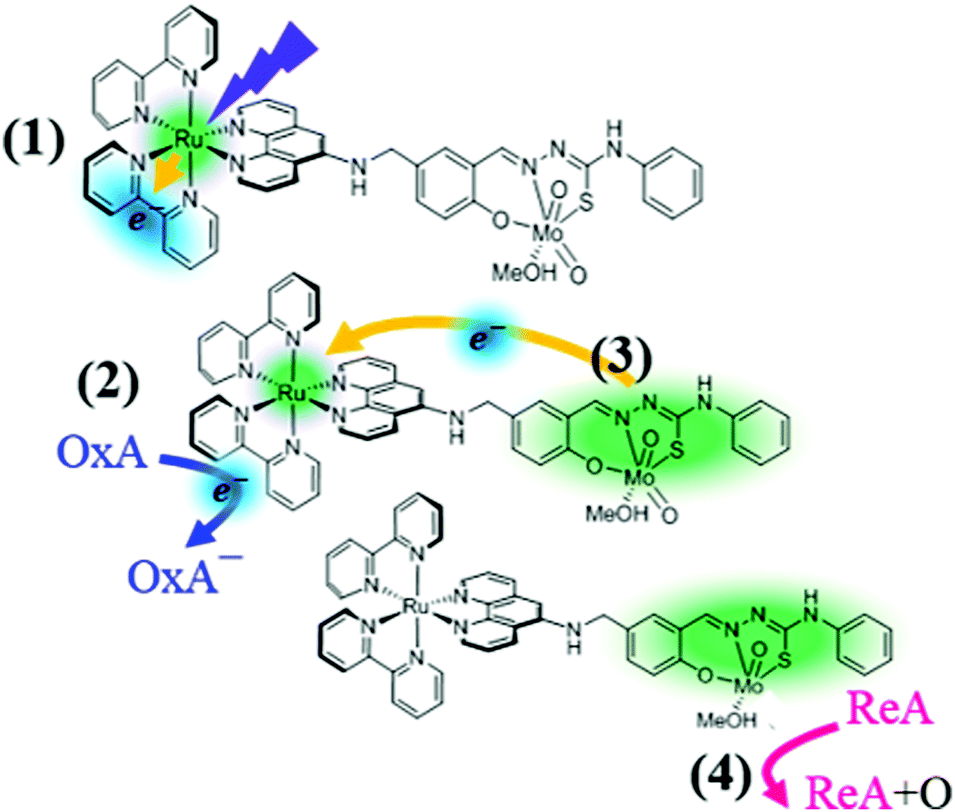 | ||
| Fig. 1 Photo-induced oxidation-enhancing (PIOE) effect in a Ru(II)–Mo(VI) dyad [Ru(bpy)2(L)MoO2(MeOH)]2+, as proposed in ref. 17: (1) the Ru(II) is photo-oxidized to Ru(III) and one of the bpy is reduced upon excitation to the lowest metal-to-ligand charge transfer excited state; (2) the photo-reduced bpy− is oxidized by a diffusion-limited reaction with an oxidizing agent (OxA), (3) the Mo(VI) unit is activated by the back-reduction of Ru(III) to Ru(II); (4) a reducing agent (ReA) is oxidized via oxygen atom transfer (OAT) from the activated cis-dioxo molybdenum unit. | ||
Since the oxidative quenching process of the photo-excited bpy− is diffusion-limited, the production of a highly reactive one-electron oxidized Mo(VI) unit is expected to happen on μs to ms timescale.
It is worth noting that the formal oxidation state of the molybdenum is already 6+ and cannot increase further. Thus, the eT following the oxidative quenching is most likely from the non-innocent phenol-thiosemicarbazone ligand system on the catalytic molybdenum unit, towards the photo-oxidized Ru(III) unit. As this kinetic model was proposed based on catalytic studies and steady-state measurements, a further validation with time-resolved techniques is highly desirable. With this motivation we carried out femtosecond (fs) transient absorption (TA) studies of the Ru(II)–Mo(VI) dyad upon photoexcitation of the Metal-to-Ligand Charge Transfer (MLCT) states of the ruthenium unit (for experimental details see ESI†). Additionally, we investigated the reference system [Ru(bpy)2(phen-NH2)]2+ (see ESI† for the molecular structures) to disentangle the dynamics of Ru(III)–bpy−/phen− MLCT states from those of the catalytic molybdenum unit, and ultimately to identify the initial steps of the PIOE process (see ESI†).
Fig. 2A shows a representative selection of TA spectra of the Ru(II)–Mo(VI) dyad. We observed, immediately after excitation, the characteristic signatures of a Ru(II)-to-bpy MLCT transition.18 In addition, at λprobe > 500 nm we found a picosecond (ps) dynamics which is absent in the reference (Fig. S2, ESI†). To quantify the timescale of these dynamics and to disentangle the spectral contributions from each dyad moiety, we did global analysis using singular value decomposition; the outcome (decay associated spectra, DASs, and lifetimes, τ) is reported in Fig. 2B (fittings of kinetics at representative wavelengths are shown in Fig. S3, ESI†).
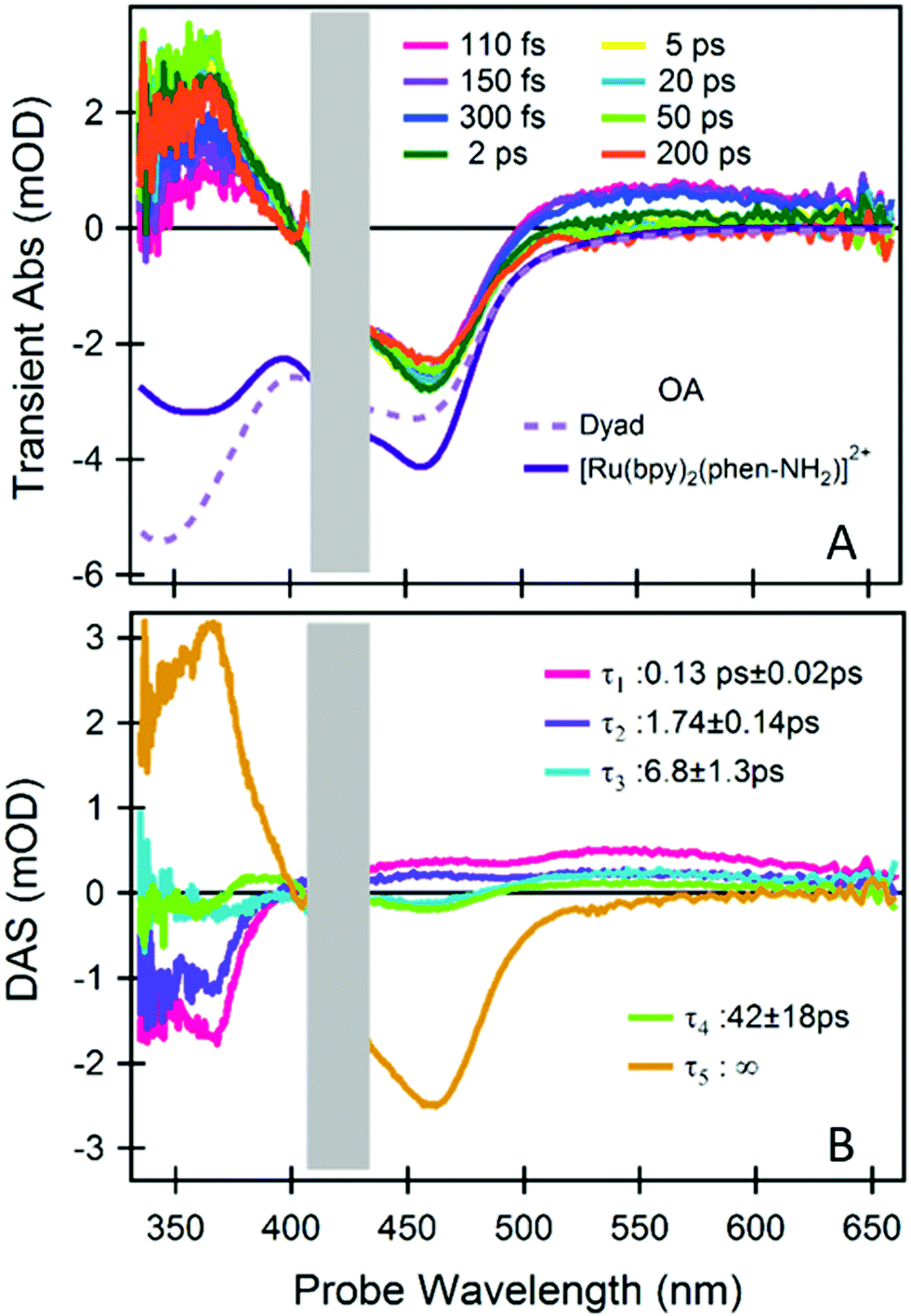 | ||
| Fig. 2 fs-TA spectroscopy of the dyad. (A) A representative selection of TA spectra upon photoexcitation at 425 nm. The inverted steady-state optical absorption (OA) of the dyad and of the reference [Ru(bpy)2(phen-NH2)]2+ are also shown. (B) Time-spectrum decomposition analysis, decay associated spectra (DASs) and the relevant decay time constants are reported. The spectral range contaminated by pump scattering is masked by a grey box. TA measurements and analysis of the reference compound are reported in the ESI.† | ||
The comparison with the reference system [Ru(bpy)2(phen-NH2)]2+ (see ESI†) indicates that both the Ru(III)–phen− and the Ru(III)–bpy− MLCT states are excited at 425 nm and that the former is followed by a biphasic ligand-to-ligand charge transfer from the phen− to one of the neutral bpy in 130 fs and 1.74 ps (DASτ1 and DASτ2). This implies that the final excited state of the photoactive ruthenium unit is a [Ru(III)(bpy−)(bpy)(phen)]2+ state, regardless of the excited MLCT transition and therefore of the excitation wavelength within the lowest OA band. The long-lived ground state bleach (GSB) signal at 450 nm in Fig. 2A and the DASτ5 with τ5 ∼ ∞ reveals that this state, and accordingly the charge density on the bpy− ligand, stays populated well beyond the investigated time window (0–200 ps), as also observed in the reference (see ESI†).
The dynamics at λprobe > 500 nm is distinctive of the dyad, and therefore of the catalytic Mo(VI) unit and the bridging ligand. We can isolate their spectral signature by subtracting the spectrum obtained at the earliest time (200 fs), which primarily contains the signals from the photoactive ruthenium unit only (Fig. 3A).
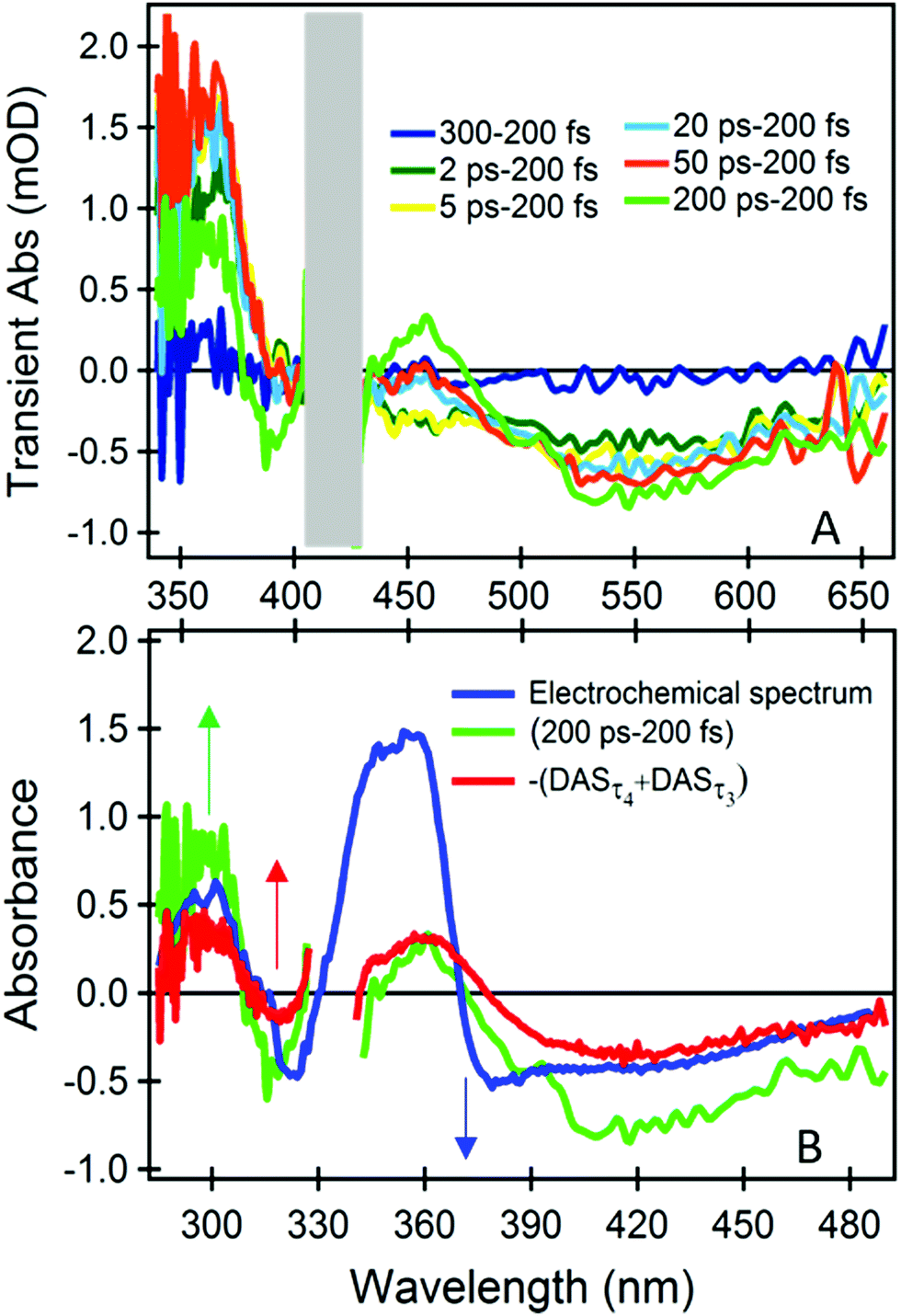 | ||
| Fig. 3 Fig. 3 (A) Difference TA spectra with respect to 200 fs TA at different time delays. (B) Comparison of the spectrum from spectro-electrochemical oxidation of the corresponding molybdenum complex [MoO2(LMe)MeOH]17 with 200 ps–200 fs differential TA spectrum and the inverse sum of the ps components from global analysis –(DASτ4 + DASτ3). To compare the two set of spectra a relative shift of 0.65 eV was applied in the energy domain (Fig. S7, ESI†). | ||
The difference TA spectrum “200 ps–200 fs” and the difference spectrum obtained during spectro-electrochemical oxidation of the catalytic control compound ([MoO2(LMe)MeOH]),17 resemble each other closely and over an extended spectral range, after applying a shift of 0.65 eV (Fig. 3B). This spectral shift is justified since the global charge of the electrochemically oxidized control compound [MoO2(LMe)MeOH] and the photoexcited dyad are different. Therefore, we can infer that (i) the early signals at λprobe > 500 nm originate from transitions of the neutral catalytic Mo(VI) unit, which are red-shifted by the strong electrostatic field of the dipole photo-induced on the Ru moiety,19,20 and (ii) the ps dynamics are due to an oxidative process of the chromophoric part of the Mo unit, i.e. the conjugated phenolate-thiosemicarbazone ligand.21 The latter is due to a decrease of the charge on the ligand, induced by the photooxidized Ru(III) in its proximity. Based on electrostatic argumentations we could tentatively relate the corresponding reductive process to acceptor orbitals on the non-innocent ligand close to the bridge, or on the bridge itself. However, the molecule is not necessarily as flat and stretched as in Fig. 1 since the linker is very flexible, and a folded structure is possible. Therefore, we cannot exclude a (partial) localization on the cis-dioxo Mo(VI) unit or on ruthenium unit, but it does not cause any detectable distortion of the associated TA spectra.
Also the inverted sum of the ps components from the global analysis –(DASτ3 + DASτ4) is similar at any wavelength (Fig. 3B). Hence, DASτ3 and DASτ4 in Fig. 2B describe the rise of a signal related to the oxidation dynamics of the Mo(VI)-coordinated chromophoric ligand. As the DASτ2 component shows the same behaviour at λprobe > 500 nm, although still dominated by the phen− → bpy− eT (see also Fig. S7 (ESI†) and respective discussion), we can infer that these oxidation dynamics are multi-exponential and spanning from 2 to 60 ps (see also Fig. S3C, ESI†). In addition, we performed the same measurements and analysis but upon 450 nm excitation to rule out that the observed dynamics could occur due to a direct excitation of the molybdenum unit, since at 450 nm we excite the photoactive unit even more selectively (Fig. S6A, ESI†). The results are identical regardless of the excitation wavelength (see Fig. S7 and respective discussion, ESI†).
Thus we can conclude that upon photoexcitation of the MLCT transitions of the photoactive ruthenium unit (regardless if Ru → bpy or Ru → phen) we observe the spectroscopic signature of the oxidation of the catalytic Mo(VI) unit (specifically of the coordinated non-innocent phenol-thiosemicarbazone ligand), which rises with a distribution of rates spanning from 2 to 60 ps, with ca. half of the eT occurring within 2 ps. The quantum yield of this process is unitary as proven by the complete bleaching of the signature of the neutral Mo(VI) unit at λprobe > 500 nm (Fig. 2, Fig. S5, ESI†).
In conclusion, our study reveals that the initial step of the PIOE effect in a molybdenum oxotransferase model Ru(II)–Mo(VI) dyad is due to an oxidative process of the chromophoric part of the Mo unit, i.e. the conjugated phenolate-thiosemicarbazone ligand, that occurs immediately after photo-oxidation of Ru(II) to Ru(III). This oxidative process spans from few ps to tens of ps and leads to the catalytic activation of the Mo(VI) moiety. These dynamics are not accompanied by any recovery of the ground state population of the photoactive unit, meaning that the dynamics in the Ru(II) and Mo(VI) units are not coupled within our time window. We can also explain why the lifetime of the bi-activated dyad is ∼1 μs:17 since the lifetime of 3MLCT states in [Ru(bpy)3]2+ is ∼1 μs,22 whereas the subsequent back eT toward the Mo(VI) unit should happen on similar timescales as the forward one (i.e. sub-ns), we expect that the lifetime of the bi-activated dyad is ultimately determined by the Ru(III)–bpy− 3MLCT relaxation.
Hence, we propose a revised mechanism of the PIOE effect, which is sketched in Fig. 4. Beside ruling out any diffusion-limited process, the simultaneous presence of both photo-activated states in the Ru(III) and the Mo(VI) unit revealed an unexpected scenario where both photo-activated sites coexist in the same system. Moreover, the charge from the Mo(VI) coordination sphere is not transferred to the ruthenium unit but rather to the acceptor orbitals close to the bridging ligand or on the cis-dioxo Mo(VI) unit.
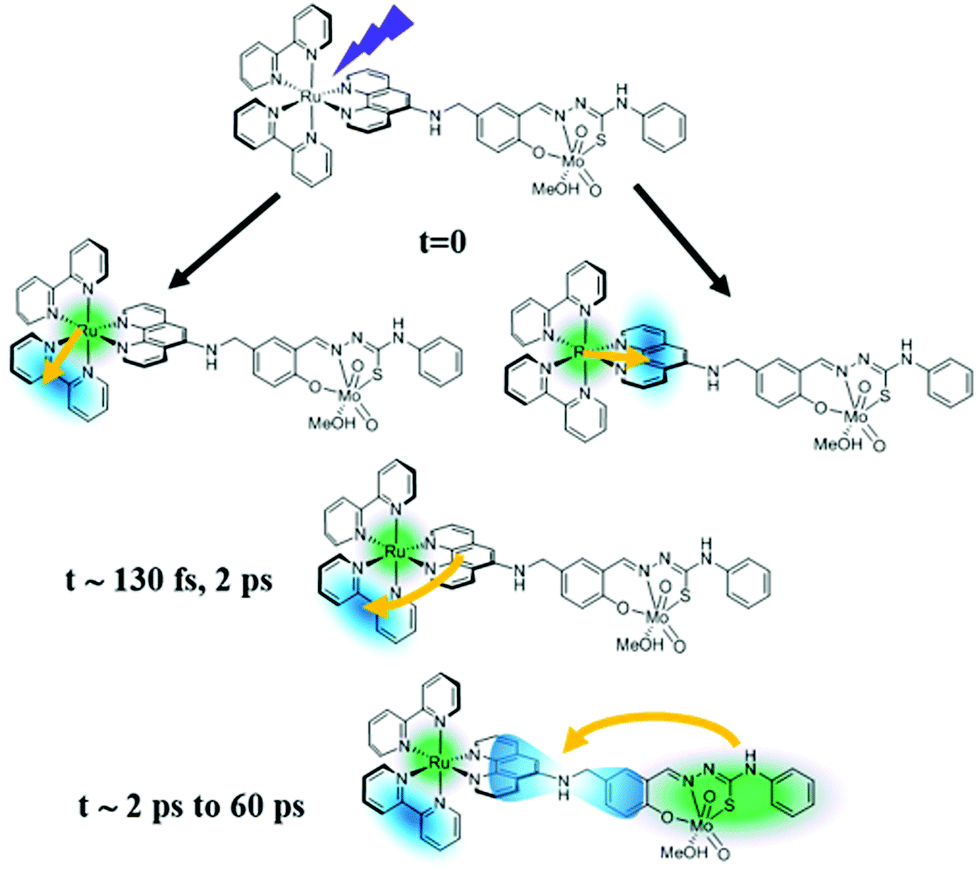 | ||
| Fig. 4 Proposed mechanism for the initial steps leading to the photo-induced activation of the catalytic molybdenum-containing unit. | ||
This novel and ultrafast photocatalytic trigger on such a small supramolecular dyad system has not been reported before and therefore it will have an important impact on modelling photocatalytic processes in bimetallic dyads, not only regarding the fs to ps range, but also on longer timescales, such as μs and even ms to s, namely on timescales longer than the expected lifetime of the bi-activated dyad. For instance, one of the first implications is that both the neutralization of the bpy− ligand by an oxidizing agent and the reduction of the catalytic molybdenum-based unit by a reducing agent can potentially proceed in parallel (Fig. S8, ESI†). On the other hand, the OAT reactions of Mo complexes can be very slow (even seconds to minutes23), while the oxidative quenching by MV2+ of the bpy− ligand is mainly diffusion-limited (typically μs). Indeed, in the experiments using MV2+ and PPh3 as oxidizing and reducing agents, respectively, the catalytic rate enhancement was observed only upon irradiation in the presence of MV2+ and the OAT to the PPh3 was an extremely slow process (∼ minutes).17 This points to the conclusion that the oxidative quenching by MV2+ prolongs the lifetime of the oxidized Mo unit beyond μs timescale, preventing the charge recombination at the Mo unit before the very slow OAT process occurs. Conversely, faster catalytic events at the Mo unit could lead to a different reaction path. Remarkably, despite the slow catalytic activity at the Mo unit cannot be directly correlated to the ultrafast dynamical processes bringing the systems to the bi-activated state, the formation of this state and the dynamical processes at the Ru units are crucial for the catalytic function.
While this study revealed the formation of a photo-activated bi-reactive state and its dynamics, it raises yet more questions relating to the role of the solvent or specific nuclear coordinates in modulating the sequence of eT events, and to the electronic and structural changes involved in the process. In particular, how the charge of the Ru(III) and Mo(VI) units differ at early times, and how the local geometry of the non-innocent ligand in the Mo(VI) unit changes. In perspective it will be crucial to carry out a study with structure sensitive techniques, such as ultrafast X-ray and polarization-resolved optical spectroscopies.
The following financial supports are acknowledged: ERC Starting Grant 279599-FunctionalDyna and Swiss NSF (grant 200021_172696) by MNHP and AC; The NCCR MUST of the Swiss NSF by AC, TF and ER; the EPSRC (EP/J019666/1) by AKDK; International Max Planck Research School for Ultrafast Imaging and Structural Dynamics (IMPRS-UFAST) and the European XFEL by TKC; Comunidad de Madrid and Universidad Autónoma de Madrid through Beatriz Galindo research project (SI2/PBG/2020-00003), Spanish MICIU through R&D project (PID2019-108678GB-I00), IMDEA-Nanociencia through Severo Ochoa Programme in R&D (SEV-2016-0686) and Polish NCN through SONATA BIS 6 grant (2016/22/E/ST4/00543) by WG. AKDK thanks Prof. R. N. Perutz for helpful discussions and Dr. N. Jasim for experimental support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Harel, Y. Bromberg, P. G. Falkowski and D. Bhattacharya, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7042–7047 CrossRef CAS PubMed.
- J. J. Warren and J. M. Mayer, Biochemistry, 2015, 54, 1863–1878 CrossRef CAS PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Chem. Rev., 2010, 110, 7024–7039 CrossRef CAS PubMed.
- S. Ghosh, G. Hogarth, N. Hollingsworth, K. B. Holt, S. E. Kabir and B. E. Sanchez, Chem. Commun., 2014, 50, 945–947 RSC.
- J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2011, 4, 26–30 CrossRef PubMed.
- A. Thapper, S. Styring, G. Saracco, A. W. Rutherford, B. Robert, A. Magnuson, W. Lubitz, A. Llobet, P. Kurz, A. Holzwarth, S. Fiechter, H. de Groot, S. Campagna, A. Braun, H. Bercegol and V. Artero, Green, 2013, 3, 43–57 Search PubMed.
- C. Herrero, A. Quaranta, W. Leibl, A. W. Rutherford and A. Aukauloo, Energy Environ. Sci., 2011, 4, 2353–2365 RSC.
- S. Ott, M. Borgström, M. Kritikos, R. Lomoth, J. Bergquist, B. Åkermark, L. Hammarström and L. Sun, Inorg. Chem., 2004, 43, 4683–4692 CrossRef CAS PubMed.
- P. A. Summers, J. A. Calladine, F. Ghiotto, J. Dawson, X.-Z. Sun, M. L. Hamilton, M. Towrie, E. S. Davies, J. McMaster, M. W. George and M. Schröder, Inorg. Chem., 2016, 55, 527–536 CrossRef PubMed.
- X. Lang, J. Zhao and X. Chen, Chem. Soc. Rev., 2016, 45, 3026–3038 RSC.
- R. A. Angnes, Z. Li, C. R. D. Correia and G. B. Hammond, Org. Biomol. Chem., 2015, 13, 9152–9167 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. Macmillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- W. Iali, P.-H. Lanoe, S. Torelli, D. Jouvenot, F. Loiseau, C. Lebrun, O. Hamelin and S. Ménage, Angew. Chem., Int. Ed., 2015, 54, 8415–8419 CrossRef CAS PubMed.
- M. H. Wall, P. Basu, T. Buranda, B. S. Wicks, E. W. Findsen, M. Ondrias, J. H. Enemark and M. L. Kirk, Inorg. Chem., 1997, 36, 5676–5677 CrossRef CAS PubMed.
- P. Basu, A. M. Raitsimring, M. J. LaBarre, I. K. Dhawan, J. L. Weibrecht and J. H. Enemark, J. Am. Chem. Soc., 1994, 116, 7166–7176 CrossRef CAS.
- A. Ducrot, B. Scattergood, B. Coulson, R. N. Perutz and A.-K. Duhme-Klair, Eur. J. Inorg. Chem., 2015, 3562–3571 CrossRef CAS.
- A. B. Ducrot, B. A. Coulson, R. N. Perutz and A.-K. Duhme-Klair, Inorg. Chem., 2016, 55, 12583–12594 CrossRef CAS PubMed.
- A. N. Tarnovsky, W. Gawelda, M. Johnson, C. Bressler and M. Chergui, J. Phys. Chem. B, 2006, 110, 26497–26505 CrossRef CAS PubMed.
- M. Koch, M. Myahkostupov, D. G. Oblinsky, S. Wang, S. Garakyaraghi, F. N. Castellano and G. D. Scholes, J. Am. Chem. Soc., 2017, 139, 5530–5537 CrossRef CAS PubMed.
- M. Meister, B. Baumeier, N. Pschirer, R. Sens, I. Bruder, F. Laquai, D. Andrienko and I. A. Howard, J. Phys. Chem. C, 2013, 117, 9171–9177 CrossRef CAS.
- B. L. Souza, L. A. Faustino, F. S. Prado, R. N. Sampaio, P. I. S. Maia, A. E. H. Machado and A. O. T. Patrocinio, Dalton Trans., 2020, 49, 16368–16379 RSC.
- A. Cannizzo, F. van Mourik, W. Gawelda, G. Zgrablic, C. Bressler and M. Chergui, Angew. Chem., Int. Ed., 2006, 45, 3174–3176 CrossRef CAS PubMed.
- R. H. Holm, Chem. Rev., 1987, 87, 1401–1449 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc00750e |
| This journal is © The Royal Society of Chemistry 2021 |