
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid 18F-labeling via Pd-catalyzed S-arylation in aqueous medium†
Swen
Humpert‡
 a,
Mohamed A.
Omrane‡
ab,
Elizaveta A.
Urusova
ab,
Lothar
Gremer
cd,
Dieter
Willbold
cd,
Heike
Endepols
abe,
Raisa N.
Krasikova
fg,
Bernd
Neumaier
*ab and
Boris D.
Zlatopolskiy
ab
a,
Mohamed A.
Omrane‡
ab,
Elizaveta A.
Urusova
ab,
Lothar
Gremer
cd,
Dieter
Willbold
cd,
Heike
Endepols
abe,
Raisa N.
Krasikova
fg,
Bernd
Neumaier
*ab and
Boris D.
Zlatopolskiy
ab
aInstitute of Neuroscience and Medicine, Nuclear Chemistry (INM-5), Forschungszentrum Jülich GmbH, Jülich 52428, Germany. E-mail: b.neumaier@fz-juelich.de
bInstitute of Radiochemistry and Experimental Molecular Imaging, University Hospital Cologne, Cologne 50931, Germany
cInstitute of Biological Information Processing, Structural Biochemistry (IBI-7), Forschungszentrum Jülich, Jülich 52425, Germany
dInstitut für Physikalische Biologie, Heinrich-Heine-Universität Düsseldorf, Düsseldorf 40225, Germany
eDepartment of Nuclear Medicine, University Hospital Cologne, Cologne 50937, Germany
fN. P. Bechtereva Institute of the Human Brain, St.-Petersburg 197376, Russia
gSt.-Petersburg State University, St.-Petersburg 199034, Russia
First published on 15th March 2021
Abstract
We report radiolabeling of thiol-containing substrates via Pd-catalyzed S-arylation with 2-[18F]fluoro-5-iodopyridine, which is readily accessible using the “minimalist” radiofluorination method. The practicality of the procedure was confirmed by preparation of a novel PSMA-specific PET-tracer as well as labeling of glutathione, Aβ oligomer-binding RD2 peptide, bovine serum albumin and PSMA I&S.
Since its introduction in the 1970s, positron emission tomography (PET) and hybrid methods like PET/CT and PET/MRI have become indispensable tools for high precision medicine and drug development.1,2 This imaging technique utilizes probes labeled with positron-emitting radionuclides (PET-tracers), which interact specifically with a molecular target or biochemical process of interest. Fluorine-18 remains the most popular PET radionuclide owing to its favorable decay properties and good accessibility in the form of [18F]fluoride. However, further PET developments necessitate easy access to emerging and established PET-tracers. A plethora of 18F-labeling techniques have been established.3,4 In particular, recently developed late-stage radiofluorination methods such as transition metal-mediated 18F-fluorination have significantly simplified the accessibility of numerous PET probes.5,6 Emerging approaches based on Al–F, B–F and Si–F chemistry enabled in many cases direct access to 18F-fluorinated peptides.7 The [18F]AlF protocol was also used for direct radiolabeling of proteins.8 In contrast, the conditions required for other radiofluorination methods are incompatible with sensitive compounds and biomolecules like peptides, proteins and nucleic acids. Furthermore, synthesis of radiolabeling precursors can be challenging, so that direct 18F-labeling is not always well suited for high throughput production of libraries of radiolabeled compounds. In this case, a modular (indirect) approach that starts with the preparation of a radiolabeled building block bearing a reactive group is more preferable.9 In a second step, this prosthetic group is coupled to an appropriate functionality of the molecule to be labeled. Whereas amino and thiol reactive building blocks are conventionally used for 18F-fluorination of peptides and proteins, tryptophan or tyrosine residues have recently been targeted as well.10 While most biopolymers contain numerous amino groups (e.g. ω-NH2 of Lys residues), the number of SH-groups (e.g. Cys moieties) seldom exceeds 1–2 per molecule, enabling conjugation with higher site selectivity. As thiol-selective prosthetic groups, 18F-labeled maleimides, vinyl sulfones, phenyloxadiazolyl methylsulfones, and the Umemoto reagent have been used.11–16 Among them, S-conjugation of maleimides affords radiolabeled thioethers as mixtures of two stereoisomers, which are not always sufficiently stable under physiological conditions.17 Vinyl sulfones18 lack complete selectivity towards S- vs. N-nucleophiles and are less reactive than maleimides. Furthermore, the preparation of these prosthetic groups usually involves multiple steps and subsequent HPLC isolation, increasing synthesis time, diminishing radiochemical yields (RCYs) and preventing automation. Recently, Al Shuaeeb et al. disclosed a fast Pd-catalysed S-arylation of Cys-containing molecules with various (hetero)aryl halides in aqueous medium at ambient temperature.19 The aim of the present study was to transfer this approach into PET-chemistry.
4-[18F]Fluoroiodobenzene ([18F]FIB) was developed for indirect radiolabeling via Pd-catalyzed cross coupling reactions.20 However, [18F]FIB prepared from respective aryl-λ3-iodane21 or sulfonium precursors22 must be purified by HPLC to remove aryl iodide side products. Furthermore, in our hands, RCYs of [18F]FIB did not exceed 30%. In search of a more practical alternative to [18F]FIB, we turned our attention to the hitherto unknown building block 5-iodo-2-[18F]fluoropyridine ([18F]1).
[18F]1 was produced from easily accessible DABCO-substituted precursor 2, which was in turn prepared in two steps from the commercially available 5-iodo-2-hydroxypyridine (3) according to the procedure of Richard et al.23 (Scheme 1 and ESI†), using the “minimalist” protocol.21,24 To this end, [18F]fluoride was eluted from an anion exchange resin with a solution of 2 in MeOH. After evaporation of MeOH, the residue was taken up in DMSO and the reaction mixture was heated at 100 °C for 15 min.25 [18F]1 could be isolated by solid phase extraction (SPE) in RCYs of 77 ± 7% (n >10) with excellent radiochemical purity (>99%) and reasonable molar activity (29 GBq μmol−1 for 1.37 GBq of [18F]1) within 25–30 min.
 | ||
| Scheme 1 Synthesis of 5-iodo-2-[18F]fluoropyridine ([18F]1) and its conjugation with thiols 4, 6, 9, 12, 14 and 16. Conditions: (a) Tf2O, Py, 0 °C to r.t., 16 h; (b) DABCO, THF, 0 °C, 5 days, 70–81% over two steps; (c) (i) elution of [18F]F− (0.03–5 GBq) with 2 in MeOH, (ii) evaporation of MeOH, 80 °C, 10 min, (iii) DMSO, 100 °C, 15 min followed by RP SPE, 77 ± 7%; (d) XantPhos Pd G3, r.t. – 37 °C, 1–6 min. RD-2-Cys: all-D-tridecapeptide: H-p-t-l-h-t-h-n-r5-c-NH2. BSAreduced: bovine serum albumin partially reduced by TCEP. PSMA I&S: MA-s3-y-nal-k[Sub-K-CO-E]-OH. TCEP: tris(2-carboxyethyl)phosphine; MA: mercaptoacetyl; nal: (R)-(2-naphthyl)alanyl; Sub: suberoyl (1,8-octanedioyl); K-CO-E: Lys-ureido-Glu. | ||
The applicability of [18F]1 for radiolabeling via S-arylation using the protocol of Al-Shuaeeb et al.19 was initially assessed using Boc-Cys-OMe (4) as a model thiol (Scheme 1). Short incubation (6 min) of [18F]1 with the substrate (2–6 μmol) in THF (0.24 mL) using XantPhos Pd G3 catalyst (2–24 mol%) and Et3N (3 equiv.) as base at ambient temperature afforded the 18F-labeled protected amino acid [18F]5 in RCYs§ of >90% (Table S1, ESI†).
Next, application of the procedure for peptide labeling was evaluated. Accordingly, reduced L-glutathione (H2N-γ-Glu-Cys-Gly-CO2H; GSH; 6) was conjugated with [18F]1 in 50% THF at ambient temperature using XantPhos Pd G3 (2–8 mol%) and Et3N (3 equiv.), furnishing [18F]7 in >90% RCY§ (Table S1, ESI†). In these experiments, 3 μmol of thiol 6 was applied, which is a substrate amount suitable for labeling of small molecules including short peptides. However, it is unacceptably high in the case of larger peptides and proteins including antibody fragments and constructs thereof, as separation of labeled products from remaining precursors can be difficult or even impossible. High precursor content in PET-tracers can affect PET image quality and cause adverse effects in patients. Consequently, optimization of the different parameters of the conjugation step with the aim to achieve reasonable RCYs at lower thiol loading was carried out, first, using aliquots of [18F]1 (Table S1, ESI†). Whereas with 2 μmol GSH almost quantitative labeling was achieved, further reduction of precursor amount led to a high variability of RCYs and at GSH amounts below 500 nmol to a sharp decline of RCYs. We hypothesized that under the basic reaction conditions, the thiol precursor could be rapidly consumed by oxidation to the corresponding disulphide (oxidized glutathione).26 In order to prevent oxidation, the conjugation reaction was carried out under nearly neutral conditions using 0.1 M Na phosphate buffer (PB; pH = 6.7–7.8) instead of Et3N. Furthermore, different organic solvents like MeOH, MeCN and DMF were evaluated (Table S1, ESI†). Under optimized reaction conditions >70% RCYs§ were obtained with only 33 nmol 6. With 167 nmol 6 almost quantitative formation of [18F]7 in 50% MeCN was observed already after 1 min reaction time.
Having optimized the novel protocol in small-scale experiments, we turned to the study of its applicability for PET tracer production on a practical scale. Accordingly, S-arylation of 6 with [18F]1 was reevaluated using whole batches of the SPE purified radiolabeled building block (Fig. 1 and Table S2, ESI†). In this case, a reciprocal influence of precursor 2 amounts on RCYs of [18F]1 and [18F]7 was observed. While higher precursor loading afforded [18F]1 in higher RCYs, it also increased the amount of nonradioactive impurities, presumably mainly 2-[4-(5-iodopyridin-2-yl)piperazin-1-yl]ethanol, which could not be removed by SPE and interfered with the consecutive conjugation step in the case of low GSH amounts (see Fig. 1B). Nevertheless under optimized conjugation conditions [Pd cat. (0.5 mg), 50% MeCN in PB (pH = 7.3; 0.6 mL), r.t., 6 min] and [18F]1 prepared from 1.0 mg DABCO salt 2, well reproducible conjugation RCYs§ of 80–90% and 20–30% were obtained using 1 μmol or 250 nmol 6, respectively (Fig. 1B and Table S2, ESI†). Further reduction of the GSH amount was possible if [18F]1 isolated by HPLC [RCY = 69 ± 4% (n >10)] was used.
 | ||
| Fig. 1 (A) Dependency of RCYs of [18F]1 (blue) and [18F]7 (over two steps, black) as well as RCY§ of the Pd-catalyzed S-arylation of GSH (6) with [18F]1 (red) on the amount of 2 used for preparation of [18F]1. (B) Dependency of RCY of [18F]7 (over two steps, black) and RCY§ of the Pd-catalyzed S-arylation of GSH (6) with [18F]1 (RCY only for the conjugation step, red) on the amount of 6 used for the S-arylation step. RCY of [18F]1 (blue) is also shown. Conditions: experiments were carried out according to GP2 (see Chapter 3.2.2. in ESI†) using 1 μmol 6 for the conjugation step (A) or 1 mg 2 for the preparation of [18F]1 (B). All experiments were carried out at least in triplicate. | ||
Being interested in the development of novel PET probes for prostate carcinoma (PCa) imaging,27,28 we applied the procedure for the preparation of a prostate specific membrane antigen (PSMA) specific tracer [18F]8. The corresponding thiol precursor 9 was synthesized from Fmoc-Cys(MMt)-OH (10) as follows (Scheme 2 and ESI†): Fmoc-Cys(MMt)-OH was esterified using tert-butyl 2,2,2-trichloroacetamidate followed by N-Fmoc-deprotection with Et2NH in CH2Cl2 to afford H-Cys(MMt)-OtBu (11), which was subsequently allowed to react with crude PhO(CO)-Glu(OtBu)2 prepared from phenyl chloroformate and HCl·H-Glu(OtBu)2 to give the corresponding protected urea. Finally, deprotection furnished 9 in 26% over four steps. The conjugation reaction between SPE-purified [18F]1 and 9 (20 μmol), performed in 50% MeCN in PB (pH 7.3; 0.6 mL) using XantPhos Pd G3 (0.5 mg) and Et3N to adjust the pH of the reaction mixture to ca. 8.0, afforded after HPLC purification and formulation [18F]8 as a ready to use solution in RCYs of 55 ± 3% (n = 6) over two steps and within 90–100 min (Table 1, entry 1). Molar activity of [18F]8 was determined to be 11–78 GBq μmol−1 (for 0.015–1.84 GBq; n = 4). Pd content in the final tracer solution amounted to 6.4 ng/batch (ICP-MS) and was well below any level of concern.29 [18F]8 was examined by μPET imaging in healthy rats, using PSMA-expressing peripheral ganglia as surrogates for small PSMA-positive tumor lesions (Fig. 2).27,30
 | ||
Scheme 2 Preparation of precursor 9. Conditions: (a) CCl3(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)OtBu, BF3·Et2O, CH2Cl2/cyclohexane (1 NH)OtBu, BF3·Et2O, CH2Cl2/cyclohexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), 3 h, 63%; (b) Et2NH, CH2Cl2, 3 h, 89%; (c) 11, PhO(CO)-Glu(OtBu)2 [prepared from HCl·H-Glu(OtBu)2: PhO(CO)Cl, Py, THF, 0 °C–r.t., 2 h], DMSO, 60 °C, 16 h, 66%; (d) TFA/H2O/TIS (90:5:5), 2 h, 70%. :3), 3 h, 63%; (b) Et2NH, CH2Cl2, 3 h, 89%; (c) 11, PhO(CO)-Glu(OtBu)2 [prepared from HCl·H-Glu(OtBu)2: PhO(CO)Cl, Py, THF, 0 °C–r.t., 2 h], DMSO, 60 °C, 16 h, 66%; (d) TFA/H2O/TIS (90:5:5), 2 h, 70%. | ||
| Entry | Substrate | Amount | Product | RCY ± SDe,f [%] (n) |
|---|---|---|---|---|
|
a Reaction solvent: entry 1:50% MeCN in PB (600 μL; pH adjusted by Et3N to 8); entries 2 and 3:50% MeCN in PB (440 μL); entry 4:25% MeCN in BB (400 μL); 50% MeCN in PB (600 μL).
b XantPhos Pd G3: entries 1–3, 5:0.5 mg; entry 4:75 μg.
c
T (t): entries 1 and 5: r.t. (6 min); entries 2 and 3: r.t. (12 min); entry 4:37 °C (20 min).
d Entries 1, 3, 4, 5: SPE purified [18F]1; entry 2: HPLC-purified [18F]1.
e Isolated yields after HPLC (entries 1–3, 5) or GPC (entry 4).
f Number of runs.
g Reduced with TCEP. PB: 0.1 M Na phosphate buffer (pH 7.3); BB: 0.1 M Na borate buffer (pH 8.0). RCY: radiochemical yield over two steps. SD: standard deviation. TCEP: tris(2-carboxyethyl)phosphine.
|
||||
| 1 , , , | Cys-CO-Glu (9) | 20 μmol | [18F]8 | 55 ± 3 (6) |
| 2 , , , | RD2c (10) | 300 nmol | [18F]11 | 40 ± 7 (7) |
| 3 , , , | RD2c (12) | 3 μmol | [18F]13 | 41 ± 5 (3) |
| 4 , , , | BSA (14)g | 6 nmol | [18F]15 | 40 ± 9 (3) |
| 5 , , , | PSMA I&S (16) | 0.5 μmol | [18F]17 | 10 ± 3 (3) |
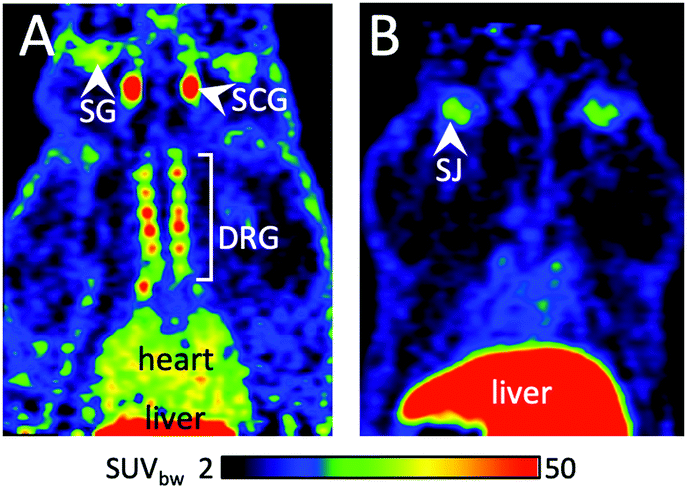 | ||
| Fig. 2 Biodistribution of [18F]8 in a healthy rat determined by μPET. (A): Horizontal view of a summed image taken 60–120 min p.i. (B): Same as A, only with the PSMA blocking agent 2-PMPA (23 mg kg−1). Note that preferential enrichment of [18F]8 is observed in the PSMA-positive ganglia with a signal-to-noise ratio of 25 in the SCG (1.5 with 2-PMPA). Liver uptake had a SUVbw of 143 (155 with 2-PMPA). Abbreviations: DRG: dorsal root ganglia; SCG: superior cervical ganglion; SG: salivary gland; SJ: shoulder joint. | ||
[18F]8 enabled the delineation of the superior cervical and dorsal root ganglia with excellent signal-to-noise ratio. The PSMA specificity of [18F]8 accumulation in ganglia was confirmed by inhibition experiments (Fig. 2). Low bone radioactivity uptake indicated high stability of the tracer to in vivo defluorination.
Next, we applied the novel protocol for the preparation of a radiolabeled analog of the brain-penetrating all-D arginine-rich dodecapeptide RD2 (H-ptlhthnr5-NH2).31 RD2 binds specifically to amyloid beta (Aβ) and was developed for the treatment of Alzheimer's disease through elimination of toxic Aβ42 oligomers. Consequently, radiofluorinated RD2 analogs could potentially be applied for Aβ-PET-imaging. An additional D-Cys residue was introduced at the C-terminus of RD2 and the resulting peptide 12 (300 nmol) was conjugated with HPLC purified [18F]1 affording after HPLC isolation and formulation the radiolabeled RD2 conjugate [18F]13 in 40 ± 7% (n = 7) RCY over two steps (Table 1, entry 2). [18F]13 was also prepared in 41 ± 5% RCY (n = 3) using the SPE purified [18F]1 and higher amounts of 12 (3 μmol) (Table 1, entry 3) with a molar activity amounting to 16–100 GBq μmol−1 (for 0.21–1.29 GBq; n = 4).
Next, the protocol was applied for 18F-fluorination of small proteins using bovine serum albumin (BSA, 14) as a model substrate. This structurally well characterized protein contains one Cys residue and 17 disulfide bonds. Native BSA (6 nmol) was partially reduced using TCEP·HCl (10 equiv.) at 37 °C for 1 h and directly conjugated to [18F]1. After 20 min incubation at 37 °C, radiolabeled BSA [18F]15 was isolated by gel permeation chromatography (GPC) in RCYs of 40 ± 9% (n = 3) (Table 1, entry 4). No formation of [18F]15 was observed in the absence of the Pd catalyst. If unreduced BSA was used, [18F]15 was isolated in 12 ± 2% RCY (n = 3) (Table S3, ESI†).
Planar scintigraphy and single photon emission tomography (SPECT) using 99mTc-labeled tracers is still an important application in nuclear medicine with more than 25 million scans worldwide per year.32,33 Several approaches can be used for 99mTc labeling of peptides. One of them consists of chelation with conjugates containing N-terminal thioacetyl or a suitably positioned Cys residue.34 Consequently, S-arylation should enable direct transformation of SPECT radiolabeling precursors into PET-tracers, so to speak a “99mTc/18F switch”. In order to examine the practicability of this approach, we 18F-fluorinated PSMA I&S (16), a precursor of the 99mTc-labeled PSMA-targeting ligand used for radioguided surgery of prostate tumors.35,36 [18F]17 was successfully prepared using [18F]1 and 500 nmol PSMA I&S in a RCY of 10 ± 3% (n = 3) over two steps (Table S2, ESI† entry 5).
In summary, Pd-catalyzed S-arylation with easily accessible 5-iodo-2-[18F]fluoropyridine is a convenient method for thiol-selective radiolabeling. Rapid kinetics under mild aqueous conditions makes it well-suited for Cys-specific 18F-labeling of peptides and proteins. The method could also be applied for the preparation of PET-tracers from conjugates originally designed for 99mTc labeling. Noteworthy, PSMA-specific [18F]8 prepared by this novel protocol turned out to be a promising candidate for prostate carcinoma imaging.
This work was supported by the DFG grant ZL 65/4-1 and RFBR grant No. 20-53-12030\20. The authors thank Dr. H. Frauendorf and G. Udvarnoki (University of Göttingen), Prof. M. Schäfer and M. Neihs (University of Cologne) for the measurement of mass spectra.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. Brady, M. L. Taylor, M. Haynes, M. Whitaker, A. Mullen, L. Clews, M. Partridge, R. J. Hicks and J. V. Trapp, Australas. Phys. Eng. Sci. Med., 2008, 31, 90–109 CrossRef CAS PubMed.
- P. M. Matthews, E. A. Rabiner, J. Passchier and R. N. Gunn, Br. J. Clin. Pharmacol., 2012, 73, 175–186 CrossRef CAS PubMed.
- S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed.
- O. Jacobson, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef CAS PubMed.
- M. Zhang, S. Li, H. Zhang and H. Xu, Eur. J. Med. Chem., 2020, 205, 112629 CrossRef CAS PubMed.
- J. Ermert, M. Benešová, V. Hugenberg, V. Gupta, I. Spahn, H.-J. Pietzsch, C. Liolios and K. Kopka, Clinical Nuclear Medicine, Springer International Publishing, Cham, 2020, pp. 49–191 Search PubMed.
- V. Bernard-Gauthier, M. L. Lepage, B. Waengler, J. J. Bailey, S. H. Liang, D. M. Perrin, N. Vasdev and R. Schirrmacher, J. Nucl. Med., 2018, 59, 568–572 CrossRef CAS PubMed.
- F. Cleeren, J. Lecina, M. Ahamed, G. Raes, N. Devoogdt, V. Caveliers, P. McQuade, D. J. Rubins, W. Li, A. Verbruggen, C. Xavier and G. Bormans, Theranostics, 2017, 7, 2924–2939 CrossRef CAS PubMed.
- R. Schirrmacher, B. Wängler, J. Bailey, V. Bernard-Gauthier, E. Schirrmacher and C. Wängler, Semin. Nucl. Med., 2017, 47, 474–492 CrossRef PubMed.
- C. W. Kee, O. Tack, F. Guibbal, T. C. Wilson, P. G. Isenegger, M. Imiołek, S. Verhoog, M. Tilby, G. Boscutti, S. Ashworth, J. Chupin, R. Kashani, A. W. J. Poh, J. K. Sosabowski, S. Macholl, C. Plisson, B. Cornelissen, M. C. Willis, J. Passchier, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 1180–1185 CrossRef CAS PubMed.
- W. Cai, X. Zhang, Y. Wu and X. Chen, J. Nucl. Med., 2006, 47, 1172–1180 CAS.
- D. O. Kiesewetter, O. Jacobson, L. Lang and X. Chen, Appl. Radiat. Isot., 2011, 69, 410–414 CrossRef CAS PubMed.
- B. de Bruin, B. Kuhnast, F. Hinnen, L. Yaouancq, M. Amessou, L. Johannes, A. Samson, R. Boisgard, B. Tavitian and F. Dollé, Bioconjugate Chem., 2005, 16, 406–420 CrossRef CAS PubMed.
- M. Cavani, D. Bier, M. Holschbach and H. H. Coenen, J. Labelled Compd. Radiopharm., 2017, 60, 87–92 CrossRef CAS PubMed.
- A. Chiotellis, F. Sladojevich, L. Mu, A. Müller Herde, I. E. Valverde, V. Tolmachev, R. Schibli, S. M. Ametamey and T. L. Mindt, Chem. Commun., 2016, 52, 6083–6086 RSC.
- S. Verhoog, C. W. Kee, Y. Wang, T. Khotavivattana, T. C. Wilson, V. Kersemans, S. Smart, M. Tredwell, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2018, 140, 1572–1575 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- T. Zhang, J. Cai, H. Wang, M. Wang, H. Yuan, Z. Wu, X. Ma and Z. Li, Bioconjugate Chem., 2020, 31, 2482–2487 CrossRef CAS PubMed.
- R. A. A. Al-Shuaeeb, S. Kolodych, O. Koniev, S. Delacroix, S. Erb, S. Nicolaÿ, J.-C. Cintrat, J.-D. Brion, S. Cianférani, M. Alami, A. Wagner and S. Messaoudi, Chem. – A Eur. J., 2016, 22, 11365–11370 CrossRef CAS PubMed.
- J. Way, V. Bouvet and F. Wuest, Curr. Org. Chem., 2013, 17, 2138–2152 CrossRef CAS.
- F. Kügler, J. Ermert, P. Kaufholz and H. Coenen, Molecules, 2014, 20, 470–486 CrossRef PubMed.
- J. D. Way and F. Wuest, J. Labelled Compd. Radiopharm., 2014, 57, 104–109 CrossRef CAS PubMed.
- M. Richard, S. Specklin, M. Roche, F. Hinnen and B. Kuhnast, Chem. Commun., 2020, 56, 2507–2510 RSC.
- B. D. Zlatopolskiy, J. Zischler, P. Krapf, R. Richarz, K. Lauchner and B. Neumaier, J. Labelled Compd. Radiopharm., 2019, 62, 404–410 CrossRef CAS PubMed.
- R. Richarz, P. Krapf, F. Zarrad, E. A. Urusova, B. Neumaier and B. D. Zlatopolskiy, Org. Biomol. Chem., 2014, 12, 8094–8099 RSC.
- R. Stevens, L. Stevens and N. Price, Biochem. Educ., 1983, 11, 70 CrossRef CAS.
- B. D. Zlatopolskiy, H. Endepols, P. Krapf, M. Guliyev, E. A. Urusova, R. Richarz, M. Hohberg, M. Dietlein, A. Drzezga and B. Neumaier, J. Nucl. Med., 2019, 60, 817–823 CrossRef CAS PubMed.
- A. Morgenroth, E. A. Urusova, C. Dinger, E. Al-Momani, T. Kull, G. Glatting, H. Frauendorf, O. Jahn, F. M. Mottaghy, S. N. Reske and B. D. Zlatopolskiy, Chem. – A Eur. J., 2011, 17, 10144–10150 CrossRef CAS PubMed.
- 29 ICH guideline Q3D (R1) on elemental impurities, 2021, https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-32.pdf.
- H. Endepols, A. Morgenroth, B. D. Zlatopolskiy, P. Krapf, J. Zischler, R. Richarz, S. Muñoz Vásquez, B. Neumaier and F. M. Mottaghy, BMC Cancer, 2019, 19, 633 CrossRef PubMed.
- T. van Groen, S. Schemmert, O. Brener, L. Gremer, T. Ziehm, M. Tusche, L. Nagel-Steger, I. Kadish, E. Schartmann, A. Elfgen, D. Jürgens, A. Willuweit, J. Kutzsche and D. Willbold, Sci. Rep., 2017, 7, 16275 CrossRef PubMed.
- National Research Council, Medical Isotope Production Without Highly Enriched Uranium, ed. C. Whipple and S. Larson, National Academies Press, Washington, D.C., 2009 DOI:10.17226/12569.
- I. Amato, Chem. Eng. News, 2009, 87, 58–64 CrossRef.
- D. Papagiannopoulou, J. Labelled Compd. Radiopharm., 2017, 60, 502–520 CrossRef CAS PubMed.
- P. Werner, C. Neumann, M. Eiber, H. J. Wester and M. Schottelius, EJNMMI Res., 2020, 10, 45 CrossRef CAS PubMed.
- S. Robu, M. Schottelius, M. Eiber, T. Maurer, J. Gschwend, M. Schwaiger and H.-J. Wester, J. Nucl. Med., 2017, 58, 235–242 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc00745a |
| ‡ Contributed equally. |
| § Determined by HPLC analysis of the crude reaction mixture after addition of H2O (radioactivity in solution ≥95%). |
| This journal is © The Royal Society of Chemistry 2021 |