
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid detection of free and bound toxins using molecularly imprinted silica/graphene oxide hybrids†
Antonio
Ruiz-Gonzalez
 a,
Adam J.
Clancy
ab and
Kwang-Leong
Choy
*a
a,
Adam J.
Clancy
ab and
Kwang-Leong
Choy
*a
aUCL Institute for Materials Discovery, UCL, 20 Gordon Street, London, WC1H0AJ, UK. E-mail: k.choy@ucl.ac.uk
bDepartment of Chemistry, UCL, London, WC1H 0AJ, UK
First published on 2nd March 2021
Abstract
Rapid, selective detection of biological analytes is necessary for early diagnosis, but is often complicated by the analytes being bound to proteins and the lack of fast and reliable systems available for their direct assessment. Here, a cheap, easily-assembled molecularly imprinted silica/graphene oxide hybrid is developed, which can selectively detect toxins linked to early-stage chronic kidney disease, down to femtomolar concentrations within 5 minutes. The hybrid material is capable of simultaneously and separately measuring free and bound analytes using with an ultra-low limit of detection in the femtomolar range, and uses processes intrinsically adaptable to any charged molecular analyte.
Detection of metabolites and molecular toxins is a key clinical tool for the early diagnosis of a large range of conditions. However, detection is often complicated by the analytes binding to proteins, becoming inaccessible for classic quantification methods which rely on free, isolated molecules. An important archetypal metabolite is indoxyl sulfate (IS); a uraemic cardiotoxin generated by the metabolism of dietary L-tryptophan.1 The accumulation of IS has been extensively reported in patients with chronic kidney disease2 and its presence is correlated to a poor prognosis of such patients.3 However, IS mostly (>90%) binds to proteins in the serum such as albumin,4 complicating analysis which would provide a vital first step towards effective monitoring and improved early treatment of patients with chronic kidney disease.
Currently, the main IS detection method5 involves initial addition of a binding competitor (e.g. sodium octanoate) to displace protein-bound IS, followed by subsequent quantification of free IS through high-performance liquid chromatography (HPLC).6 Alternative recently-developed methods include using acetonitrile as a precipitation agent before injecting the samples in the HPLC columns,7 but in all cases, these methods require a laborious preparation, and are performed over the hours timescale so are unsuitable for rapid quantification. The speed and efficacy of any test is vital to facilitate incorporation into real-world settings to better diagnose and assist in the treatment of patients with chronic kidney disease.2,3
The application of non-equilibrium methodologies such as pulse amperometry (PA) may circumnavigate these challenges. During PA, a short-lived current between a working and counter electrode is established by applying a voltage pulse. As a consequence, charged analytes locally concentrate around the electrodes since such voltage induces an ion flux, which improves the sensitivity of the sensors.8 PA has previously been proven to be useful for the detection of charged molecules in solution,9 and more recently Shvarev et al.10 demonstrated that PA can be adapted for the determination of free and total concentration of ions in the presence of chelating agents in solution; during the PA pulse, a depletion of ions close to the sensing film is thought to force the extraction of the complexed ions, temporarily allowing their detection (before subsequent rechelation). Thus, PA shows promise for the development of rapid detection systems that allow the simultaneous detection of free and bound charged molecules in solution. However, the availability of suitable materials that can be adapted for the specific detection of IS in solution is low. To date, only materials that allow the indirect detection, including enzymes such as sulfatase, have been developed.5 This enzymatic method involves the reaction of IS with sulfatase, releasing indoxyl molecules that can be detected by reaction with tetrazolium salts and measuring the changes in the absorbance at 280 nm. However, given the laborious preparation and the relatively bulky equipment needed, this technology cannot be applied for the fast and portable detection of IS. Thus, (nano)materials that allow the selective adsorption of IS in solution are needed to tackle the current challenges by developing electrochemical sensors.
The recent development of molecular imprinting technologies has facilitated a recent expansion in the solution sensor field, allowing the low-cost synthesis of materials that can specifically interact with one of a wide range of challenging analytes. The molecular imprinting process consists of the polymerisation or condensation of functional monomers in the presence of an analyte to generate a selective material.11 Molecularly imprinted mesoporous silica has been adapted for the detection of vancomycin, melamine, and trypsin, demonstrating the versatility of these materials for the detection of multiple compounds in biological environments electrochemically.12 However, akin to HPLC-based methodologies, the sensitivity of these electrochemical devices was poor in these biological environments when compared to other assays such as enzyme-linked immunosorbent assay.13
Here, a new PA-amplified sensor based on hybridised molecularly imprinted silica nanoparticles and exfoliated graphene oxide is developed for the detection of free and protein-bound IS in body fluids. To create the sensing material (Fig. 1a, full synthetic details provided in the ESI†), in brief, cyclodextrin was covalently attached to a silyl ether group by using an adapted literature procedure,14 which was subsequently mixed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/ethanol with N,N,N-trimethyl-1,1,1-trimethylsilyl ammonium iodide, tetraethyl orthosilicate, and IS, leading to silicon oxide formation, with quaternary amines and cyclodextrins templated around the IS. The mixture also contained exfoliated graphene oxide (synthesised using a modified Hummers’ method15) which acted as a support for the growing nanoporous silica. Exfoliated graphene oxide was chosen as a support due to its high capacitance16 and ability to transduce the potentiometric signal in ion-selective electrodes.17 Devices were assembled by depositing the sensing material as a 500 nm film through aerosol assisted chemical deposition,18 onto 50 nm thick gold electrodes sputtered on glass, followed by coating of a binding chitosan top layer. The molecularly imprinted silica nanoparticles had a diameter of ca. 58 nm and sat on the top of the graphene oxide layers, as clearly seen in SEM (Fig. 2a and b). The material had an average pore radius of 1.7 nm as measured by BET using a DFT estimation (ESI,† Fig. S5). The final device consisted of a three-electrode setup using the assembled material as working electrode, a platinum wire counter electrode, and a silver chloride reference.
:1 water/ethanol with N,N,N-trimethyl-1,1,1-trimethylsilyl ammonium iodide, tetraethyl orthosilicate, and IS, leading to silicon oxide formation, with quaternary amines and cyclodextrins templated around the IS. The mixture also contained exfoliated graphene oxide (synthesised using a modified Hummers’ method15) which acted as a support for the growing nanoporous silica. Exfoliated graphene oxide was chosen as a support due to its high capacitance16 and ability to transduce the potentiometric signal in ion-selective electrodes.17 Devices were assembled by depositing the sensing material as a 500 nm film through aerosol assisted chemical deposition,18 onto 50 nm thick gold electrodes sputtered on glass, followed by coating of a binding chitosan top layer. The molecularly imprinted silica nanoparticles had a diameter of ca. 58 nm and sat on the top of the graphene oxide layers, as clearly seen in SEM (Fig. 2a and b). The material had an average pore radius of 1.7 nm as measured by BET using a DFT estimation (ESI,† Fig. S5). The final device consisted of a three-electrode setup using the assembled material as working electrode, a platinum wire counter electrode, and a silver chloride reference.
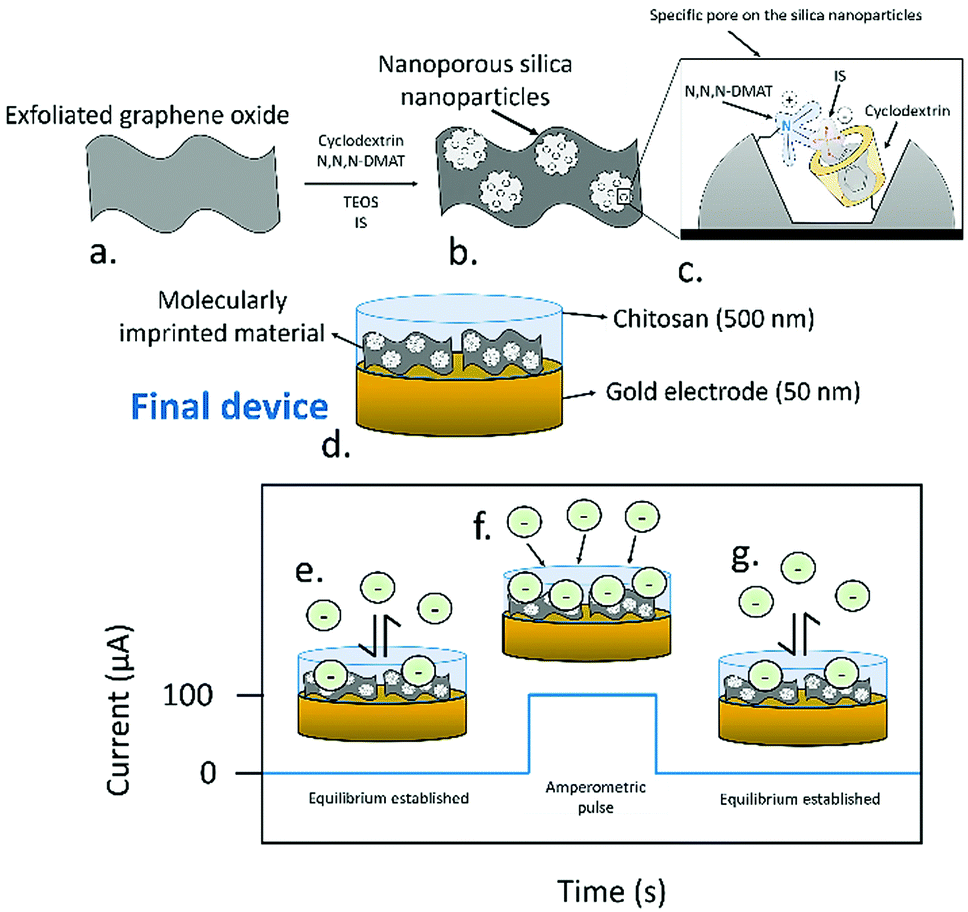 | ||
| Fig. 1 Schematic representation of the synthesis process followed for the formation of the molecular imprinted nanoparticles. (a) Exfoliated graphene oxide is used as a support for the synthesis of the selective nanoparticles. (b) Besides the target analyte (IS), used as a template, cyclodextrin bound to silyl ether groups, (N,N,N-trimethyl-3-aminopropyl) trimethoxysilane (N,N,N-DMAT) and tetraethyl orthosilicate as spacer are employed. (c) The combination of this elements generates a specific pore that allows the detection of IS in solution. (d) The final device consisted of a thin gold electrode film (50 nm) where the molecularly imprinted material was deposited, and an upper chitosan layer. The final device, including the chitosan, electrode and ionically imprinted material had a thickness of 500 nm. (e) When zero-current OCP measurements were used, an equilibrium between the IS in solution and the sensing devices was established. (f) Upon the application of an amperometric pulse, an ion flux is generated, increasing the potentiometric signal. (g) After the pulse, OCP is used again to regenerate the equilibrium conditions on the sensing electrodes. | ||
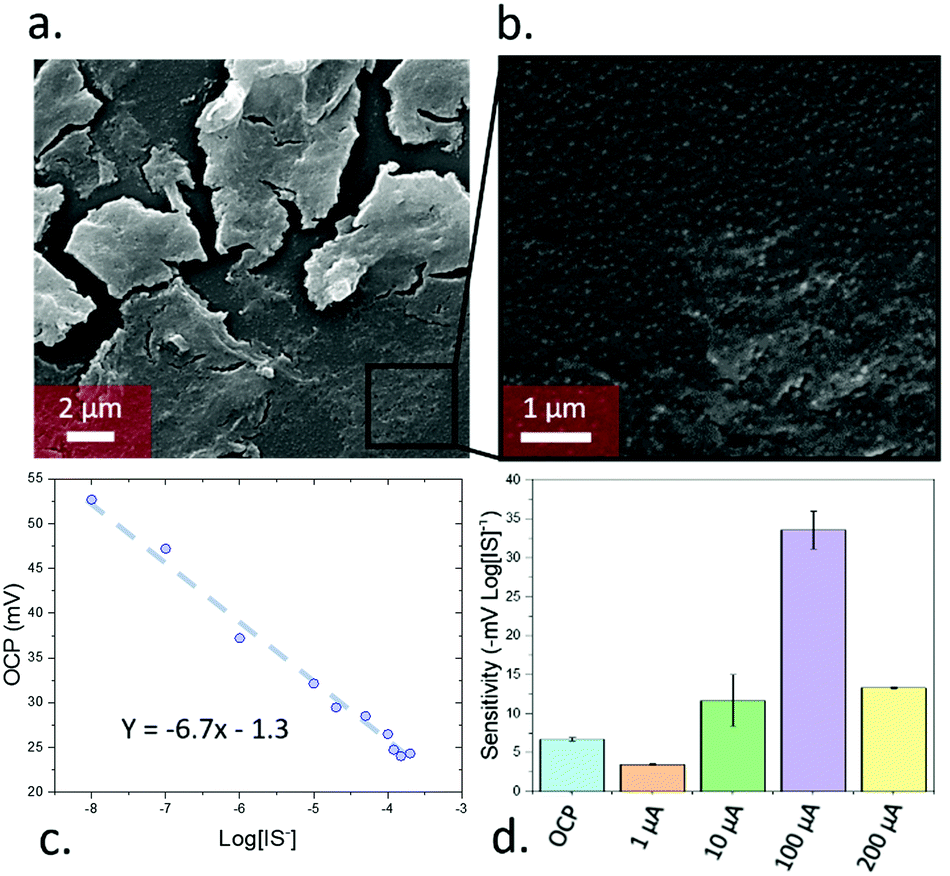 | ||
| Fig. 2 (a) nanoparticles onto the exfoliated graphene oxide. (b) Detail of the silica nanoparticles on the surface of the exfoliated silica nanoparticles. (c) Calibration plot of the sensing devices by OCP. (d) Sensitivity achieved by using pulse amperometry under 4 different current values (1, 10, 100 and 200 μA). | ||
Due to the presence of a sulfate group, IS in solution has a negative charge, allowing its quantification by measuring the potential of the electrodes. When open circuit potential was used for the measurements, the sensors operated as in the case of ion-selective electrodes. In this, case, the presence of the specific pores induced an adsorption of the IS molecules, and an equilibrium between the IS inside the sensing film and the sample was established. The transduction of the potentiometric signal was then accomplished by an electron exchange between the capacitive interlayer, in this case the graphene oxide, acting as ion transducer, and the gold contact electrodes.
To calibrate the device, the open circuit potential (OCP) was measured with IS concentrations between 10−8 to 2 × 10−4 M (the typical range found in human serum19) in simulated body fluids (ESI,† Table S1), leading to a linear relationship (Fig. 2c) of −6.7 ± 0.2 mV log[IS]−1. In addition, the limit of detection was determined by following the IUPAC guidance. Such limit of detection was calculated by measuring the OCP of the devices in the absence of IS (i.e. just simulated body fluids) and calculating the intersection between the calibration curve with this value (ESI,† Fig. S6a). This experiment was repeated 6 times and the standard deviation of the measurements was determined. This standard deviation was added to the instrument detection limit obtained by the blank measurements after multiplying it by 3. The calculated limit of detection was 2.5 × 10−15 M, one of the lowest values reported so far in the field of electrochemical sensors, and 109 times lower standard HPLC detection20 of indoxyl sulfate (ca. 10−6 M). The sensitivity achieved in this case was reproducible across different sensors in both OCP and pulse amperometric detection (ESI,† Fig. S7).
Beyond these OCP measurements, PA measurements were performed through measuring the difference immediately after a 5 s pulse of pre-set current. A 5 min OCP measurement was taken prior to the pulse to ensure stability for a total measurement time of 305 s. The use of PA initially decreased the sensitivity of the devices when using low current values (1 μA; −3.5 mV log[IS]−1), but increased for higher currents with a maximum of −34 mV log[IS]−1 seen for 100 μA (Fig. 2d). Further increases in pulse current led to a subsequent decrease in sensitivity. The device behaviour remained constant across biologically relevant pHs, degrading at extremely high or low pH values (ESI,† Fig S9a and b). Testing in high non-target ion concentrations (10 mM NaCl/KHCO3/CaCl2, 103 × [IS], ESI,† Fig S9c and d), the OCP was ion-independent and PA trends remained the same, with dampened total signal from the high dielectric environments.
When non-templated silica nanoparticles were employed for the sensing, a significantly lower sensitivity of the devices, in the range of 2.7 ± 1.6 mV Log[IS]−1 for the OCP measurements and −17.0 ± 0.3 mV Log[IS]−1 for pulse amperometry measurements, were obtained (ESI,† Fig. S8). In addition, when a control sample which did not include any silica nanoparticles were tested for the detection of IS, a low sensitivity in the range of 1.8 ± 0.2 mV Log[IS]−1 and −3 ± 2 mV Log[IS]−1 for OCP and pulse amperometry respectively were obtained. These results highlight the necessity of silica nanoparticles and a correct templating for achieving the required sensitivity of the sensing devices.
The magnitude of the applied pulse was also used to modulate the selectivity of the devices. Selectivity was studied by measuring the sensitivity of the sensors towards tryptophol, and caffeine, which show a similar structure to IS, and creatinine, a molecule commonly present in serum samples and metabolised by the kidneys, which accumulates in patients with chronic kidney disease21 (Fig. 3a). This selectivity was determined by studying the sensitivity of the devices towards these compounds by triplicates. When the OCP of the sensors was used for the calibration, the sensitivity towards caffeine, creatinine and tryptophol was relatively low (−2.7, −3.1 and −0.5 mV log[I]−1, respectively). In the case of caffeine and tryptophol, the achieved sensitivity was lower than the one obtained in the case of IS across all the tested current values. However, at 10 μA, the sensitivity towards creatinine (−14.5 mV log[I]−1) was higher than IS (−13 mV log[IS]−1), and only at 100 μA was the device more sensitive towards IS, for which the device was designed. As such, the use of an optimal current magnitude can be seen to be essential not only for ensuring a good sensitivity, but also to preserve the selectivity of the devices. Additionally, in theory, the differing response trends for differing analytes as a function of pulse current allows measurement on a solution of mixed analytes, by calibrating the sensitivity towards multiple analytes across many PA currents and measuring the total response of the mixed sample across different PA pulses.
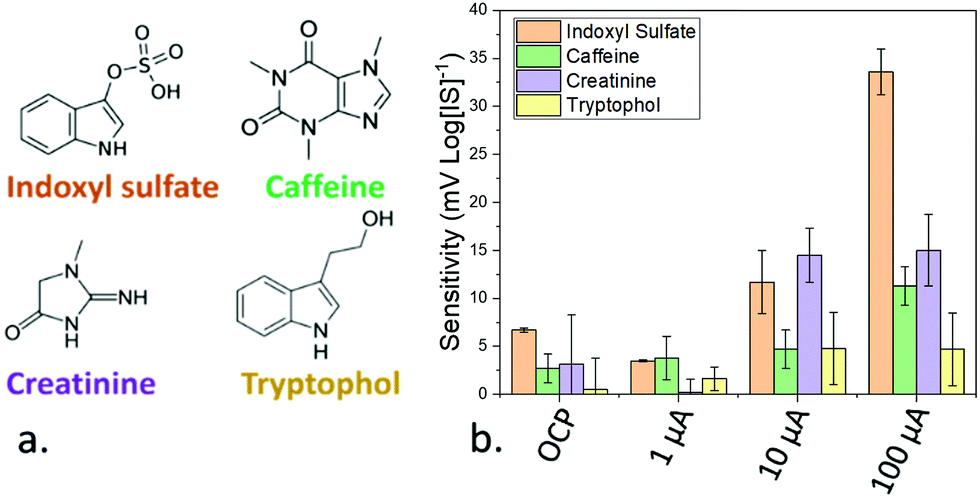 | ||
| Fig. 3 (a) Molecular structure of indoxyl sulfate and the components employed for the measurement of the selectivity of the developed molecularly imprinted silica nanomaterial. (b) Sensitivity achieved towards the components employed for testing the selectivity of the sensing device using OCP and pulse amperometry with 3 different magnitudes (1, 10 and 100 μA). The error bars represent the standard error after measuring the sensitivities by triplicates. | ||
The dependence between the observed sensitivity and selectivity of amperometric sensors is commonly observed in electrochemical sensors operating with cyclic voltammetry. The maximum current was obtained as a result of an optimal diffusion layer, due to the adsorption of the electrolytes to the sensing.22 As such, the optimal current for the adsorption of IS was 100 μA.
One of the main proposed advantages of the PA methodologies is the potential for the detection of analytes after they have been complexed or adsorbed to other molecules. Thus, pulse amperometry could be exploited for the quantification of the free and protein-bound concentration of IS in biological fluids, which is one of the major challenges in the quantification of IS. To prove the applicability of this pulse amperometry approach for the detection of free and bound IS, the sensing devices were first tested in the presence of a dispersion of activated carbon. Activated carbon presents a highly porous structure rich in polar hydroxyl and ketone groups that can adsorb organic compounds from solution,23–25 and has been employed for the removal of plasma proteins including IS previously.26 As such, it represented a perfect candidate for testing sensitivity of the device towards bound IS.
A concentration of 250 μM IS in simulated body fluids was employed (in the range of concentrations typically found in the serum of patients with chronic kidney disease19) and 10 μg mL−1 of activated carbon was added. The free and total amount of IS were determined by measuring the OCP and the response after applying a 100 μA PA pulse, respectively (Fig. 4a). The free concentration measured from the OCP was 13 μM, implying ca. 95% of the IS was absorbed onto the carbon black and incapable of transferring to the electrode surface. However, immediately after the pulse, a concentration of 249 μM was measured; approximately the concentration initially added. Similar to previous work extracting chelated ions using PA,10,27 the pulse was seen to lead to a desorption of the biologically relevant analyte from the carbon black into the solution allowing accurate quantification.
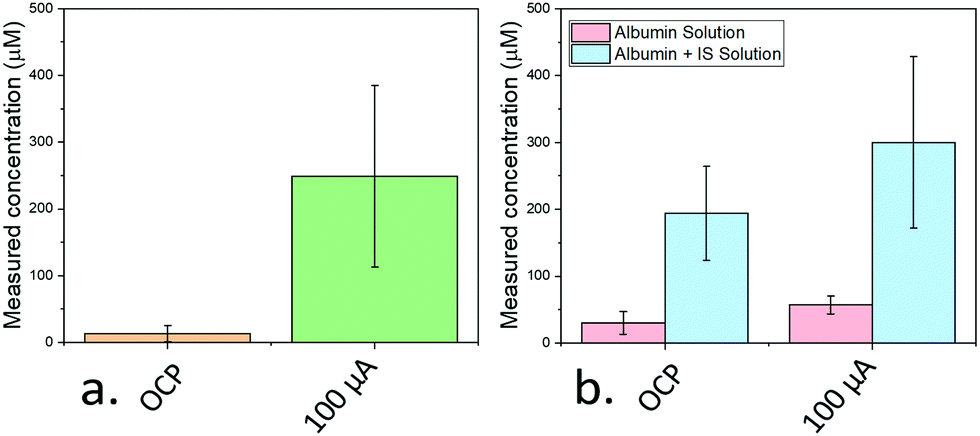 | ||
| Fig. 4 (a) Measured concentration of IS after immersing the electrodes in a solution containing 250 μM IS and 0.04 mg mL−1 activated carbon. The employed concentration is indicated by a dashed line. (b) Measured concentrations obtained after immersing the sensing devices in a solution containing 0.04 mg mL−1 albumin with and without 250 μM IS. Error bars represent standard errors. | ||
To provide an even more realistic binding environment, a 250 μM IS solution was made using SBF containing 0.04 mg mL−1 human albumin (Fig. 4b). Albumin was chosen since it represents the most common protein in serum, and the adsorption of IS to this protein has been extensively studied.28 Here, free IS in the solution measured from OCP remained high (194 μM) indicating only partial uptake of IS, while the PA (using a 1 μA pulse) measurement led to a higher value (300 μM).
The discrepancy is attributed to the presence of analytes in the original human albumin sample employed which desorb from albumin under PA, and are measured alongside the IS. Indeed, the PA-measured concentration of IS of a solution only containing albumin was 57 μM, correlating well with the measured excess above the added IS concentration (50 μM).
In summary, a sensing device based on molecularly imprinted silica has been developed for the detection of indoxyl sulfate in simulated body fluids. The material is assembled from cheap, readily available reagents and may be synthesised and fabricated as device films quickly and simply. This system could detect indoxyl sulfate into the 10−15 M range, being 109 times lower than the standard HPLC method. By using pulse amperometry, the sensitivity of the device increased by almost 6-fold with increased selectivity when tested against caffeine, creatinine and tryptophol. Additionally, the pulse amperometry could be employed for the simultaneous detection of the free and bound concentrations of IS, tested using an idealised binding support and human albumin. This test not only provides a clear route to cheap and rapid (ca. 5 min) screening for chronic kidney disease but provides a versatile framework to create a range of effective sensors using alternative biological analytes, even those typically bound to macromolecules in real-world environments.
AJC would like to thank the Society of Chemical Industry and The Ramsay Memorial Trust for funding.
Conflicts of interest
There are no conflicts to declare.References
- E. Banoglu, G. G. Jha and R. S. King, Eur. J. Drug Metab. Pharmacokinet., 2001, 26, 235–240 CrossRef CAS PubMed.
- W.-C. Liu, Y. Tomino and K.-C. Lu, Toxins, 2018, 10, 367 Search PubMed.
- X. Tan, X. Cao, J. Zou, B. Shen, X. Zhang, Z. Liu, W. Lv, J. Teng and X. Ding, Hemodialysis international. International Symposium on Home Hemodialysis, 2017, 21, 161–167.
- E. Devine, D. H. Krieter, M. Rüth, J. Jankovski and H.-D. Lemke, Toxins, 2014, 6, 416–429 Search PubMed.
- T. Abe, M. Onoda, T. Matsuura, J. Sugimura, W. Obara, N. Sasaki, T. Kato, K. Tatsumi and T. Maruyama, Ther. Apheresis Dial., 2020, 1–6, DOI:10.1111/1744-9987.13500.
- H. de Loor, B. K. Meijers, T. W. Meyer, B. Bammens, K. Verbeke, W. Dehaen and P. Evenepoel, J. Chromatogr. A, 2009, 1216, 4684–4688 CrossRef CAS PubMed.
- M. Al Za'abi, B. Ali and M. Al Toubi, J. Chromatogr. Sci., 2012, 51, 40–43 Search PubMed.
- M. Senda, H. Katano and M. Yamada, J. Electroanal. Chem., 1999, 475, 91–98 CrossRef.
- J. Ding, X. Wang and W. Qin, ACS Appl. Mater. Interfaces, 2013, 5, 9488–9493 CrossRef CAS PubMed.
- A. Shvarev and E. Bakker, Talanta, 2004, 63, 195–200 CrossRef CAS.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- E. Piletska, H. Yawer, F. Canfarotta, E. Moczko, K. Smolinska-Kempisty, S. S. Piletsky, A. Guerreiro, M. J. Whitcombe and S. A. Piletsky, Sci. Rep., 2017, 7, 11537 CrossRef PubMed.
- J. C. E. Odekerken, D. M. W. Logister, L. Assabre, J. J. C. Arts, G. H. I. M. Walenkamp and T. J. M. Welting, SpringerPlus, 2015, 4, 614 CrossRef PubMed.
- M.-L. Hsieh, G.-Y. Li, L.-K. Chau and Y.-S. Hon, J. Sep. Sci., 2008, 31, 1819–1827 CrossRef CAS PubMed.
- A. T. Habte and D. W. Ayele, Adv. Mater. Sci. Eng., 2019, 2019, 5058163 Search PubMed.
- B. Xu, S. Yue, Z. Sui, X. Zhang, S. Hou, G. Cao and Y. Yang, Energy Environ. Sci., 2011, 4, 2826–2830 RSC.
- S. Hu, R. Zhang and Y. Jia, Sensors, 2020, 20, 3500 CrossRef CAS.
- K. L. Choy, Prog. Mater. Sci., 2003, 48, 57–170 CrossRef CAS.
- C.-N. Lin, I. W. Wu, Y.-F. Huang, S.-Y. Peng, Y.-C. Huang and H.-C. Ning, J. Food Drug Anal., 2019, 27, 502–509 CrossRef CAS PubMed.
- M. Al Za'abi, B. Ali and M. Al Toubi, J. Chromatogr. Sci., 2013, 51, 40–43 Search PubMed.
- W. E. Mitch, V. U. Collier and M. Walser, Clin. Sci., 1980, 58, 327–335 CrossRef CAS PubMed.
- N. Elgrishi, J. Rountree, D. McCarthy, S. Rountree, T. Eisenhart and L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95(2), 197–206 CrossRef CAS.
- X. Tan, X. Cao, J. Zou, B. Shen, X. Zhang, Z. Liu, W. Lv, J. Teng and X. Ding, Hemodial. Int., 2017, 21, 161–167 CrossRef PubMed.
- P. S. Pamidimukkala and H. Soni, J. Env. Chem. Eng., 2018, 6, 3135–3149 CrossRef CAS.
- X. Liang, J. Chi and Z. Yang, Microporous Mesoporous Mater., 2018, 262, 77–88 CrossRef CAS.
- S. Yamamoto, T. Ito, M. Sato, S. Goto, J. J. Kazama, F. Gejyo and I. Narita, Blood purification, 2019, 48, 215–222 CrossRef CAS PubMed.
- A. Ceresa, E. Pretsch and E. Bakker, Anal. Chem., 2000, 72, 2050–2054 CrossRef CAS PubMed.
- T. Sakai, K. Yamasaki, T. Sako, U. Kragh-Hansen, A. Suenaga and M. Otagiri, Pharm. Res., 2001, 18, 520–524 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc00572c |
| This journal is © The Royal Society of Chemistry 2021 |