
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective methanethiol-to-olefins conversion over HSSZ-13 zeolite†
Miao
Yu
,
Nicoló
Tormene
,
Aleksei
Bolshakov
 ,
Brahim
Mezari
,
Anna
Liutkova
,
Nikolay
Kosinov
* and
Emiel J. M.
Hensen
*
,
Brahim
Mezari
,
Anna
Liutkova
,
Nikolay
Kosinov
* and
Emiel J. M.
Hensen
*
Laboratory of Inorganic Materials and Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, Het Kranenveld 14, 5600 MB, Eindhoven, The Netherlands. E-mail: n.a.kosinov@tue.nl; e.j.m.hensen@tue.nl
First published on 5th March 2021
Abstract
A methanethiol-to-olefins (MtTO) equivalent of methanol-to-olefins (MTO) chemistry is demonstrated. CH3SH can be converted to ethylene and propylene in a similar manner as CH3OH over SSZ-13 zeolite involving a hydrocarbon pool mechansim. Methylated aromatic intermediates were identified by 13C NMR analysis. Comparison of MtTO and MTO chemistry provides clues about the mechanism of C–C bond formation and catalyst deactivation.
The methanol-to-hydrocarbons (MTH) reaction1 catalyzed by zeolites represents a promising technology for the conversion of synthesis gas and CO2 to a variety of chemicals and fuels via methanol.2–4 Intensive investigations into the pathways of the MTH reaction have led to a broad acceptance of the hydrocarbon pool (HCP) mechanism.5,6 This mechanism involves organocatalytic intermediates of aromatic and olefinic nature, resulting in two catalytic cycles interconnected through hydrogen transfer reactions. The generality of the HCP chemistry is evident from similar mechanistic proposals for the zeolite-catalyzed conversion of methyl halides to higher hydrocarbons,7 conversion of furanics,8,9 and the dehydroaromatization of methane.10 The fact that similar chemistry can arise from different starting reactants prompted us to investigate methanethiol (CH3SH) substrate. Selective conversion of this industrially important molecule11 to olefins has not been demonstrated yet.
Earlier work by Chang and Silvestri indicated that CH3SH can be converted to a mixture of alkanes, heavy aromatics, dimethylsulfide and H2S on HZSM-5 at 482 °C.12 Huguet et al. reported that conversion of a diluted CH3SH feed (0.5 kPa in N2) in the temperature range of 400–550 °C in HZSM-5, HBEA, HFER, HMOR and HSAPO-34 mainly led to CH4 and H2S with minor amounts of ethane, propane, and aromatic hydrocarbons.13,14 Similar to methanol, CH3SH can also methylate aromatics, e.g. toluene to xylenes.15 The methylation of olefins by CH3SH can take place over zeolite catalysts as well, although it is significantly more difficult than methylation by CH3OH.16 Methylation of aromatics and olefins are important steps in the formation of organocatalytic intermediates in methanol-to-hydrocarbons chemistry.17 Such computational insights suggest that the conversion of CH3SH to hydrocarbons via the HCP mechanism should be possible over zeolite catalysts.
The conversion chemistry of CH3SH has been only scarcely investigated in comparison with that of CH3OH. As it has been noted that CH3SH readily decomposes into CH4 and H2S above 730 °C,18 we first established a temperature window that would disfavor this unselective gas-phase pathway. For this purpose, we studied the conversion of CH3SH in a quartz reactor with SiC particles in the temperature range of 350–550 °C (Fig. S1, ESI†), and found that selective catalytic conversion of CH3SH can only be achieved well below 500 °C where thermal decomposition is avoided. The decomposition products are methane, ethane, dimethylsulfide and heavier organosulfur compounds (Fig. S2, ESI†).
Typical zeolite catalysts that have found utility for the selective conversion of CH3OH to hydrocarbons are 10MR and 8MR zeolites.4 The 3-dimensional 10MR micropores of HZSM-5 (MFI topology) can be used to convert CH3OH to gasoline-range hydrocarbons. When light olefins are targeted, 8MR zeolites like HSSZ-13 (CHA topology) or its aluminophosphate counterpart HSAPO-34 are preferred. We used the CHA and MFI topologies as representative examples of small- and medium-pore zeolites with distinct shape selectivity properties to compare the conversion of CH3SH and CH3OH. The basic physico-chemical properties of the used zeolite catalysts are given in Fig. S3–S6 and Tables S1, S2 (ESI†).
The catalytic performance of both zeolites was evaluated at a reactant (CH3SH or CH3OH) partial pressure of 6 kPa balanced by N2 at atmospheric pressure. Based on the non-catalytic experiments with CH3SH, we selected a temperature of 450 °C for the initial activity evaluation. Fig. 1a shows the CH3SH and CH3OH conversion as a function of time on stream. Full conversion of CH3OH was achieved for HZSM-5 and HSSZ-13 during 6 h of time-on-stream. While the CH3SH conversion on HZSM-5 was low at the start of the reaction (t = 0.5 h, X = 26%), HSSZ-13 achieved a much higher initial conversion of ∼80%, which slowly decreased to ∼40% after 6 h. The hydrocarbon product distributions obtained within first 2 h on stream are given in Fig. 1b. CH3OH conversion on HZSM-5 resulted in the expected product slate dominated by light olefins and C4+ hydrocarbons and a small amount of CH4.19 CH3SH conversion on HZSM-5 gave mainly CH4 with minor amounts of C2H4 and C4+ hydrocarbons. Strikingly, whereas the product distributions for the conversion of CH3OH and CH3SH on HZSM-5 were very different, the use of HSSZ-13 offered a comparable hydrocarbon selectivity to light olefins from CH3SH and CH3OH. The CH4 selectivity on HSSZ-13 was slightly higher for CH3SH. The decreasing CH3SH conversion with time on stream for HSSZ-13 led to a decreasing yield of light olefins (Fig. 1c) and a higher overall coke selectivity. We also evaluated two other zeolites, HBEA with larger 12MR pores and HFER with interconnected 8MR and 10MR pores. Both zeolites displayed similar poor catalytic performance as HZSM-5 (Fig. S7, ESI†). Among the zeolite topologies evaluated herein, only HSSZ-13 catalyst can selectively produce light olefins. Although the product distribution obtained in the MTO and MtTO reactions over HSSZ-13 are similar, the deactivation rate in the MtTO reaction is higher in combination with a lower initial CH3SH conversion. These observations can be explained by faster formation of deactivating species and thermodynamic limitations of CH3SH conversion to olefins and H2S (90% at the applied conditions).
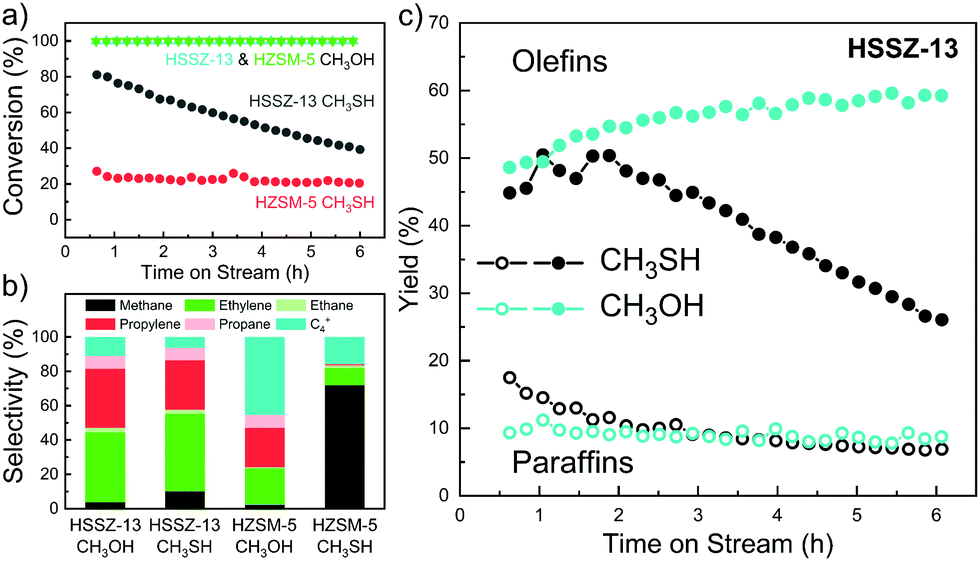 | ||
| Fig. 1 Catalytic performance of HZSM-5 and HSSZ-13 in conversion of CH3OH and CH3SH. (a) Time on stream profiles of CH3OH/CH3SH conversion and (b) carbon basis product distributions observed within first 2 h of respective reactions; (c) yield of C1–C3 paraffins and C2–C3 olefins from CH3OH and CH3SH over HSSZ-13. Conditions: 6 kPa of CH3OH/CH3SH in N2 (5 mL min−1), 450 °C, 100 mg of catalysts, 6 h. | ||
Next, we studied the influence of the reaction temperature on CH3SH conversion on HSSZ-13 (Fig. S8, ESI†). At a higher temperature of 500 °C, the unselective decomposition of CH3SH contributed substantially to the conversion, resulting in a CH4 hydrocarbon selectivity of 45%. At a lower reaction temperature of 400 °C, the reaction was characterized by an induction period of ∼2 h with a product distribution similar to the reaction at 450 °C. The catalytic results suggest that the main catalytic conversion pathways of CH3OH and CH3SH are similar for the 8MR zeolite HSSZ-13, yet differ for zeolites with larger pores. The induction period observed at a relatively low reaction temperature of 400 °C for CH3SH conversion is consistent with findings for the MTO reaction.20,21 As the MTO reaction over HSSZ-13 is known to involve the HCP mechanism, we speculate that such a pool of hydrocarbons is also present in the micropores of HSSZ-13 during the conversion of methanethiol to olefins (MtTO).
We employed 13C MAS NMR spectroscopy to study the organic reaction intermediates occluded in the micropores of the used zeolite catalysts. Direct excitation and cross polarization of 13C NMR spectra of HSSZ-13 and HZSM-5 catalysts after 6 h reaction at 450 °C and 2D 1H-13C{1H} HETCOR spectrum of the used HSSZ-13 sample are presented in Fig. 2a, Fig. S9 (ESI†) and Fig. 2b, respectively. The 13C NMR spectra feature two main groups of signals at (i) 10–30 ppm, corresponding to sp3 carbon atoms and (ii) 120–140 ppm, representing sp2 carbon atoms in arenes and olefins.22–24 These features are also observed in the 2D HETCOR spectra of the used HSSZ-13 sample. The 13C signal at 129 ppm mainly correlates with the 1H signal at 8.6 ppm, which indicates arenes to be the main sp2 carbon species.25 Considering the intensity differences, the used HSSZ-13 zeolite contained much more aromatic species than the used HZSM-5 sample, which is consistent with the known preference for the aromatic cycle on 8MR zeolites.22–24
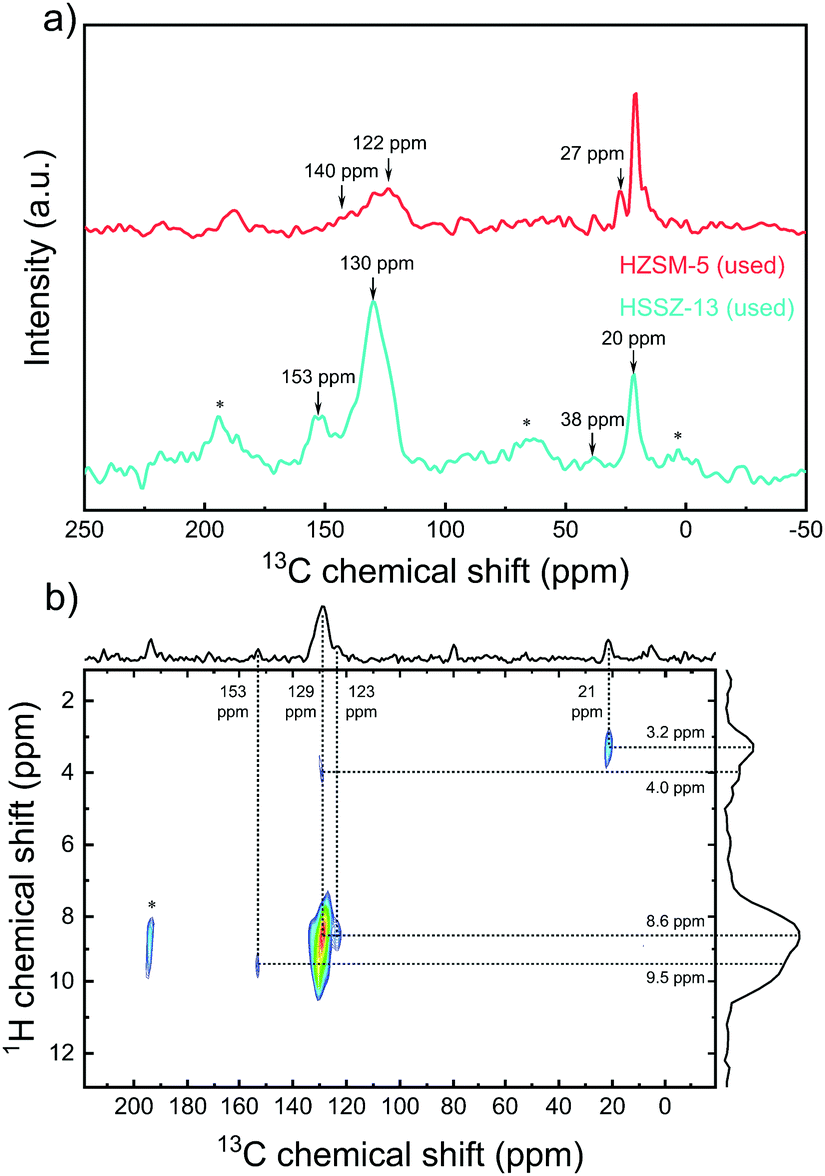 | ||
| Fig. 2 (a) Direct excitation of 13C NMR spectra of the used HZSM-5 and HSSZ-13 and (b) 1H-13C{1H} HETCOR MAS NMR 2D spectrum of used HSSZ-13, after 6 h MtTO reaction at 450 °C. Asterisks denote spinning side bands. | ||
Furthermore, the NMR spectrum of used HZSM-5 presents a higher relative intensity of sp3 carbon species (signal ∼21 ppm), implying a higher concentration of saturated hydrocarbons than typically reported during CH3OH conversion over HZSM-5.26 We hypothesize that this is due to the formation and deposition of non-volatile thiols, thioethers and oligosulfides on HZSM-5.27 Interestingly, the signal at 153 ppm in the 13C NMR spectrum of HSSZ-13, which is characteristic for polymethylcyclopentyl cations, has also been reported during CH3OH conversion on HSSZ-13 and HSAPO-34 and these species are argued to be important reaction intermediates in the pairing mechanism of the aromatic cycle.28 The NMR spectra of the catalysts used in MtTO do not contain the typical 13C NMR signals between 50 ppm to 80 ppm due to surface methoxy species and trimethyloxonium species observed during CH3OH conversion.29 Instead, signals at 38 ppm and 27 ppm were, respectively, observed on HSSZ-13 and HZSM-5. Such 13C NMR signals can be assigned to surface trialkylsulfonium cations (alkyl = methyl or ethyl), which have also been experimentally reported on HZSM-5.30 Quantum-chemical calculations demonstrated that the trimethylsulfonium cation is stable on HSSZ-13.31 It can be suggested that trialkylsulfonium might be an intermediate in MtTO in analogy with the proposed role of trimethyloxonium in MTO chemistry.29 It has actually been proposed that trimethyloxonium might be an important intermediate in the formation of the first C–C bond.29,32,33 In summary, 13C NMR spectroscopy revealed key differences between HZSM-5 and HSSZ-13 catalysts in the MtTO reaction. First, the 13C NMR spectrum of used HSSZ-13 featured a signal of polymethylcyclopentyl cations, which strongly points to the importance of the aromatic cycle of HCP mechanism. Second, while the 13C NMR spectrum of used HSSZ-13 contains similar bands as typically observed after CH3OH conversion, the corresponding spectrum of the used HZSM-5 catalyst contains more intense sp3 carbon signals, suggesting the deposition of saturated non-volatile deposits such as sulfur-containing aliphatics formed during oligomerization of CH3SH. The formation of such species in the wider HZSM-5 pores might explain the rapid deactivation of this catalyst.
Next, we characterized the used zeolites in more detail with 27Al NMR spectroscopy, TGA-MS and XPS. The 27Al NMR spectra of fresh and used catalysts in Fig. S10 (ESI†) show that dealumination did not occur during the MtTO reaction. Accordingly, the degradation of the zeolite crystals cannot be a reason for the quick deactivation. The TG profiles and normalized MS signals of CO2 (m/z = 44) and SO2 (m/z = 64) of used HSSZ-13 and HZSM-5 during heating in 20 vol% O2 in He are shown in Fig. 3a and d, respectively (data for used HBEA and HFER in Fig. S11, ESI†). The CO2 and SO2 MS signals derive from the oxidation of carbon- and sulfur-containing deposits, providing insight into the nature of the coke species deposited during the MtTO reaction. Although HZSM-5 deactivated very fast during the catalytic test and showed much lower conversion levels, the total amount of coke was only 4.7 wt% (Fig. S12, ESI†). The coke content of used HSSZ-13 was much higher (11.6 wt%) and comparable with the coke content of HSSZ-13 after use in the MTO reaction.34 TGA-MS profiles show similar CO2 peaks for the HSSZ-13 and HZSM-5 zeolites used in MtTO with a main combustion feature at 500–600 °C, which was also observed and assigned to coke in zeolite micropores formed during the MTO reaction and linked to polyolefinic and polyaromatic species (Fig. 3c).23,35 On the contrary, the SO2 profiles are different for the two used zeolites. Three contributions can be distinguished: the SO2 feature below 300 °C is likely related to the decomposition, desorption and combustion of relatively light components having boiling points lower than 300 °C, such as CH3SH, CH3SCH3, and CH3SSCH3. The high intensity of the SO2 signal in comparison with CO2 suggests that elemental sulfur is present as well. A comparison of this SO2 feature for the two used zeolites shows that HZSM-5 contains much more of such organosulfur compounds. The second SO2 peak observed in the range between 300–400 °C appears only in HZSM-5 together with a shoulder in the MS signal of CO2. Based on its relatively low combustion temperature as compared to typical coke species located inside the pores,36 we assign this peak to the combustion of heavier sulfur-containing molecules located on the external surface and in near-surface regions of zeolite crystals. Considering that HZSM-5 contains a lower total amount of coke and yet exhibits a lower activity in the MtTO reaction, we argue that these sulfur-containing deposits (Fig. 3c) cause the rapid deactivation of HZSM-5. The third SO2 peak between 500–600 °C appears together with the large CO2 peak. Accordingly, we assign this feature to the typical diffusion-limited combustion of coke species confined inside zeolite micropores.36 The total amount of sulfur-containing species in the used HSSZ-13 catalyst is much smaller than in used HZSM-5.
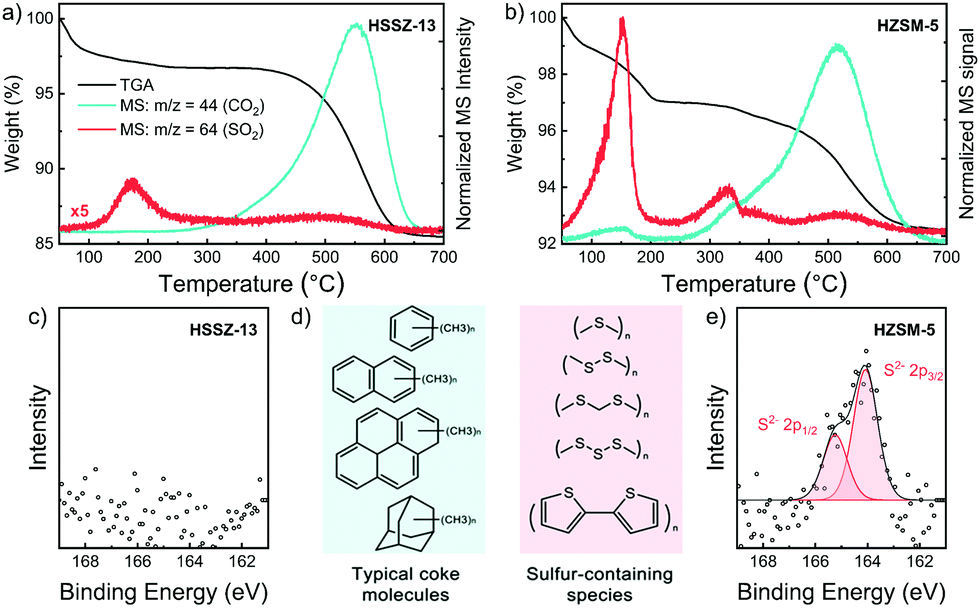 | ||
| Fig. 3 TGA curves and MS signals of CO2 (m/z = 44) and SO2 (m/z = 64) collected under air flow of used (a) HSSZ-13 and (d) HZSM-5. XP spectra of S 2p region of used (b) HSSZ-13 and (e) HZSM-5 (c) typical coke compositions in MTO reaction and potential sulfur-containing coke species in MtTO reaction. | ||
XPS spectra of used HSSZ-13 and HZSM-5 (Fig. 3b and e) underpin this conclusion. In line with the TG-MS data, no S 2p signals were detected at the surface of used HSSZ-13, whereas clear evidence for the presence of thiols and polysulfides at the surface of the used HZSM-5 zeolite was provided by XPS.37 For HSSZ-13, the S 2p XPS feature was only observed when the MtTO reaction was carried out at 500 °C (Fig. S13, ESI†). Together with the high CH4 selectivity observed under these conditions (Fig. S8b, ESI†), we conclude that formation of non-volatile sulfur compounds at the external surface is due to the unselective and non-catalytic decomposition of CH3SH to CH4. Selective conversion of CH3SH at optimal temperature of 450 °C is related to the sulfur-free HCP aromatics, confined in the SSZ-13 cages.
In conclusion, we demonstrated for the first time the selective conversion of CH3SH to light olefins and identified HSSZ-13 as a promising catalyst for this novel reaction in the temperature range of 400–450 °C. We established that the HSSZ-13 pores contain aromatic reaction intermediates that have earlier been linked to the aromatic cycle of the MTH reaction. The specific pore geometry of this zeolite (small 8MR windows connecting larger cages) allows formation of the HCP intermediates essential for the selective conversion of CH3SH to light olefins, but prevents reactions that give rise to bulky deposits that quickly deactivate the near-surface regions of zeolite crystals with larger 10MR and 12MR pores. Next, the main finding of this work that CH3SH can undergo similar chemistry as CH3OH can provide clues about the much-debated aspects of MTH chemistry such as the formation of the first C–C bond and the deactivation mechanism. Formaldehyde formed by disproportionation of CH3OH can alkylate olefinic and aromatic chain growth carriers, accelerating the relative rates of hydrogen transfer and thus the transformation of active HCP intermediates into deactivating species.38,39 Moreover, formaldehyde has been implicated in the chemistry leading to the formation of the first C–C bond.40,41 CO derived from CH3OH decomposition can be involved in Koch-type carbonylation of methoxy species or dimethylether to form the first C–C bond. While both routes are not feasible with CH3SH (i.e., carbon monosulfide CS and thioformaldehyde CH2S are not stable under the applied conditions), the presented MtTO reaction still exhibits common characteristics of MTH chemistry such as C–C bond formation and deactivation. It is therefore worthwhile to investigate these aspects in more detail. The finding that HSSZ-13 zeolite can convert CH3SH with a hydrocarbon selectivity to light olefins close to the selectivity observed for MTO opens a new research direction for optimizing 8MR zeolites towards valorization of CH3SH into valuable chemical building blocks. Further mechanistic studies, including density functional theory calculations, spectroscopy and isotope labelling, will shed light on the dominant reaction pathways of MtTO chemistry. Finally, reaction optimization including other 8MR zeotypes such as silicoaluminophosphates with milder acidity (e.g. SAPO-34) and hierarchically porous zeolites will undoubtedly lead to improved MtTO performance.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. L. Meisel, J. P. McCullough, C. H. Lechthaler and P. B. Weisz, Chem. Tech., 1976, 6, 86–89 CAS.
- X. Niu, J. Gao, Q. Miao, M. Dong, G. Wang, W. Fan, Z. Qin and J. Wang, Microporous Mesoporous Mater., 2014, 197, 252–261 CrossRef CAS.
- M. Conte, J. A. Lopez-Sanchez, Q. He, D. J. Morgan, Y. Ryabenkova, J. K. Bartley, A. F. Carley, S. H. Taylor, C. J. Kiely, K. Khalid and G. J. Hutchings, Catal. Sci. Technol., 2012, 2, 105–112 RSC.
- U. Olsbye, S. Svelle, M. Bjrgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149, 458–464 CrossRef CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1996, 161, 304–309 CrossRef CAS.
- S. Svelle, S. Aravinthan, M. Bjørgen, K. P. Lillerud, S. Kolboe, I. M. Dahl and U. Olsbye, J. Catal., 2006, 241, 243–254 CrossRef CAS.
- E. A. Uslamin, H. Saito, N. Kosinov, E. Pidko, Y. Sekine and E. J. M. Hensen, Catal. Sci. Technol., 2020, 10, 2774–2785 RSC.
- E. A. Uslamin, N. Kosinov, G. A. Filonenko, B. Mezari, E. Pidko and E. J. M. Hensen, ACS Catal., 2019, 9, 8547–8554 CrossRef CAS.
- N. Kosinov, A. S. G. Wijpkema, E. Uslamin, R. Rohling, F. J. A. G. Coumans, B. Mezari, A. Parastaev, A. S. Poryvaev, M. V. Fedin, E. A. Pidko and E. J. M. Hensen, Angew. Chem., Int. Ed., 2018, 57, 1016–1020 CrossRef CAS PubMed.
- W. Leuchtenberger, K. Huthmacher and K. Drauz, Appl. Microbiol. Biotechnol., 2005, 69, 1–8 CrossRef CAS PubMed.
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 CrossRef CAS.
- E. Huguet, B. Coq, R. Durand, C. Leroi, R. Cadours and V. Hulea, Appl. Catal., B, 2013, 134–135, 344–348 CrossRef CAS.
- V. Hulea, E. Huguet, C. Cammarano, A. Lacarriere, R. Durand, C. Leroi, R. Cadours and B. Coq, Appl. Catal., B, 2014, 144, 547–553 CrossRef CAS.
- C. Cammarano, E. Gay, A. Finiels, O. Fajula and V. Hulea, ACS Catal., 2019, 9, 605–609 CrossRef CAS.
- Q. M. Li, M. Zhang, C. M. Wang, Y. A. Zhu, X. G. Zhou and Z. K. Xie, Mol. Catal., 2018, 446, 106–114 CrossRef CAS.
- S. Svelle, U. Olsbye, F. Joensen and M. Bjørgen, J. Phys. Chem. C, 2007, 111, 17981–17984 CrossRef CAS.
- A. H. Sehon and B. D. Darwent, J. Am. Chem. Soc., 1954, 76, 4806–4810 CrossRef CAS.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef.
- E. Borodina, H. Sharbini Harun Kamaluddin, F. Meirer, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, J. Ruiz-Martinez and B. M. Weckhuysen, ACS Catal., 2017, 7, 5268–5281 CrossRef CAS PubMed.
- E. Borodina, F. Meirer, I. Lezcano-González, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, J. Ruiz-Martinez and B. M. Weckhuysen, ACS Catal., 2015, 5, 992–1003 CrossRef CAS.
- C. Wang, J. Xu and F. Deng, ChemCatChem, 2020, 12, 965–980 CrossRef CAS.
- E. Epelde, M. Ibañez, A. T. Aguayo, A. G. Gayubo, J. Bilbao and P. Castaño, Microporous Mesoporous Mater., 2014, 195, 284–293 CrossRef CAS.
- D. Fu, A. Lucini Paioni, C. Lian, O. van der Heijden, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 20024–20030 CrossRef CAS PubMed.
- W. Dai, C. Wang, M. Dyballa, G. Wu, N. Guan, L. Li, Z. Xie and M. Hunger, ACS Catal., 2015, 5, 317–326 CrossRef CAS.
- A. Dutta Chowdhury, I. Yarulina, E. Abou-Hamad, A. Gurinov and J. Gascon, Chem. Sci., 2019, 10, 8946–8954 RSC.
- F. Freeman and C. N. Angeletakis, Org. Magn. Reson., 1983, 21, 86–93 CrossRef CAS.
- S. Xu, A. Zheng, Y. Wei, J. Chen, J. Li, Y. Chu, M. Zhang, Q. Wang, Y. Zhou, J. Wang, F. Deng and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 11564–11568 CrossRef CAS PubMed.
- X. Wu, S. Xu, W. Zhang, J. Huang, J. Li, B. Yu, Y. Wei and Z. Liu, Angew. Chem., Int. Ed., 2017, 56, 9039–9043 CrossRef CAS PubMed.
- E. J. Munson, A. A. Kheir and J. F. Haw, J. Phys. Chem., 1993, 97, 7321–7327 CrossRef CAS.
- J. Baltrusaitis, T. Bučko, W. Michaels, M. Makkee and G. Mul, Appl. Catal., B, 2016, 187, 195–203 CrossRef CAS.
- E. J. Munson, A. A. Kheir, N. D. Lazo and J. F. Haw, J. Phys. Chem., 1992, 96, 7740–7746 CrossRef CAS.
- E. J. Munson and J. F. Haw, J. Am. Chem. Soc., 1991, 113, 6303–6305 CrossRef CAS.
- X. Zhu, J. P. Hofmann, B. Mezari, N. Kosinov, L. Wu, Q. Qian, B. M. Weckhuysen, S. Asahina, J. Ruiz-Martínez and E. J. M. Hensen, ACS Catal., 2016, 6, 2163–2177 CrossRef CAS.
- J. Li, G. Xiong, Z. Feng, Z. Liu, Q. Xin and C. Li, Microporous Mesoporous Mater., 2000, 39, 275–280 CrossRef CAS.
- N. Kosinov, E. A. Uslamin, F. J. A. G. Coumans, A. S. G. Wijpkema, R. Y. Rohling and E. J. M. Hensen, ACS Catal., 2018, 8, 8459–8467 CrossRef CAS PubMed.
- J. Riga and J. J. Verbist, J. Chem. Soc., Perkin Trans. 2, 1983, 1545–1551 RSC.
- A. Hwang, M. Kumar, J. D. Rimer and A. Bhan, J. Catal., 2017, 346, 154–160 CrossRef CAS.
- A. Hwang and A. Bhan, Acc. Chem. Res., 2019, 52, 2647–2656 CrossRef CAS PubMed.
- A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 15840–15845 CrossRef CAS PubMed.
- Y. Liu, S. Müller, D. Berger, J. Jelic, K. Reuter, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Angew. Chem., Int. Ed., 2016, 55, 5723–5726 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc00397f |
| This journal is © The Royal Society of Chemistry 2021 |