
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insight into intramolecular chemical structure modifications by on-surface reaction using photoemission tomography†
Iulia
Cojocariu
 *a,
Florian
Feyersinger
b,
Peter
Puschnig
b,
Luca
Schio
c,
Luca
Floreano
c,
Vitaliy
Feyer
*ad and
Claus M.
Schneider
ad
*a,
Florian
Feyersinger
b,
Peter
Puschnig
b,
Luca
Schio
c,
Luca
Floreano
c,
Vitaliy
Feyer
*ad and
Claus M.
Schneider
ad
aPeter Grünberg Institute (PGI-6), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: i.cojocariu@fz-juelich.de; v.feyer@fz-juelich.de
bInstitute of Physics, University of Graz, NAWI Graz, Universitätsplatz 5, 8010 Graz, Austria
cCNR-IOM, Lab. TASC, s.s. 14 km 163,5, 34149 Trieste, Italy
dFakultät f. Physik and Center for Nanointegration Duisburg-Essen (CENIDE), Universität Duisburg-Essen, D-47048 Duisburg, Germany
First published on 15th February 2021
Abstract
The sensitivity of photoemission tomography (PT) to directly probe single molecule on-surface intramolecular reactions will be shown here. PT application in the study of molecules possessing peripheral ligands and structural flexibility is tested on the temperature-induced dehydrogenation intramolecular reaction on Ag(100), leading from CoOEP to the final product CoTBP. Along with the ring-closure reaction, the electronic occupancy and energy level alignment of the frontier orbitals, as well as the oxidation state of the metal ion, are elucidated for both the CoOEP and CoTBP systems.
Octaethylporphyrins (OEPs) are a representative class of porphyrin molecules with peripheral ethyl substituents. During the synthesis or the in situ metalation processes, different metal ions can be incorporated into the center of the porphyrin macrocycle leading to the formation of metal-OEP (M-OEP) molecules.1 Upon anchoring to a solid substrate, molecules adapt their structural conformation to the local environment, deforming the macrocycle and/or reorienting the substituents.1 Thereby, the peripheral ethyl groups of the adsorbed OEP molecule can point away from and towards the substrate, allowing the metal ions in M-OEP to be closer to or farther from the metal substrate, respectively.
Together with the surface-driven conformational changes, the porphyrin molecules may undergo on-surface temperature-induced chemical changes of their molecular structure.2–6 A well-known surface-assisted ring-closure reaction in the case of metal-OEP involves the peripheral ethyl groups and results in the transformation of the OEP in tetrabenzoporphyrin (TBP), which occurs via the dehydrogenation of two neighbouring ethyl groups, followed by the formation of six-membered carbon rings (see Fig. 1).5,7,8 This on-surface reaction can deeply alter the intriguing properties of the porphyrin array. For instance, a substantial increase in the effective spin moment has been reported in the case of iron(II)-TBP formed upon on-surface reaction on Au(111) from iron(II)-OEP.5 In contrast to this system, CoOEP on Cu(100) has shown a sizable magnetic moment, while CoTBP displayed a quenched moment on this substrate.4 Notably, the manipulation of the magnetic anisotropy in metal porphyrin molecules induced by ring-closure reactions is a key issue for the development of molecular-based spintronic devices.
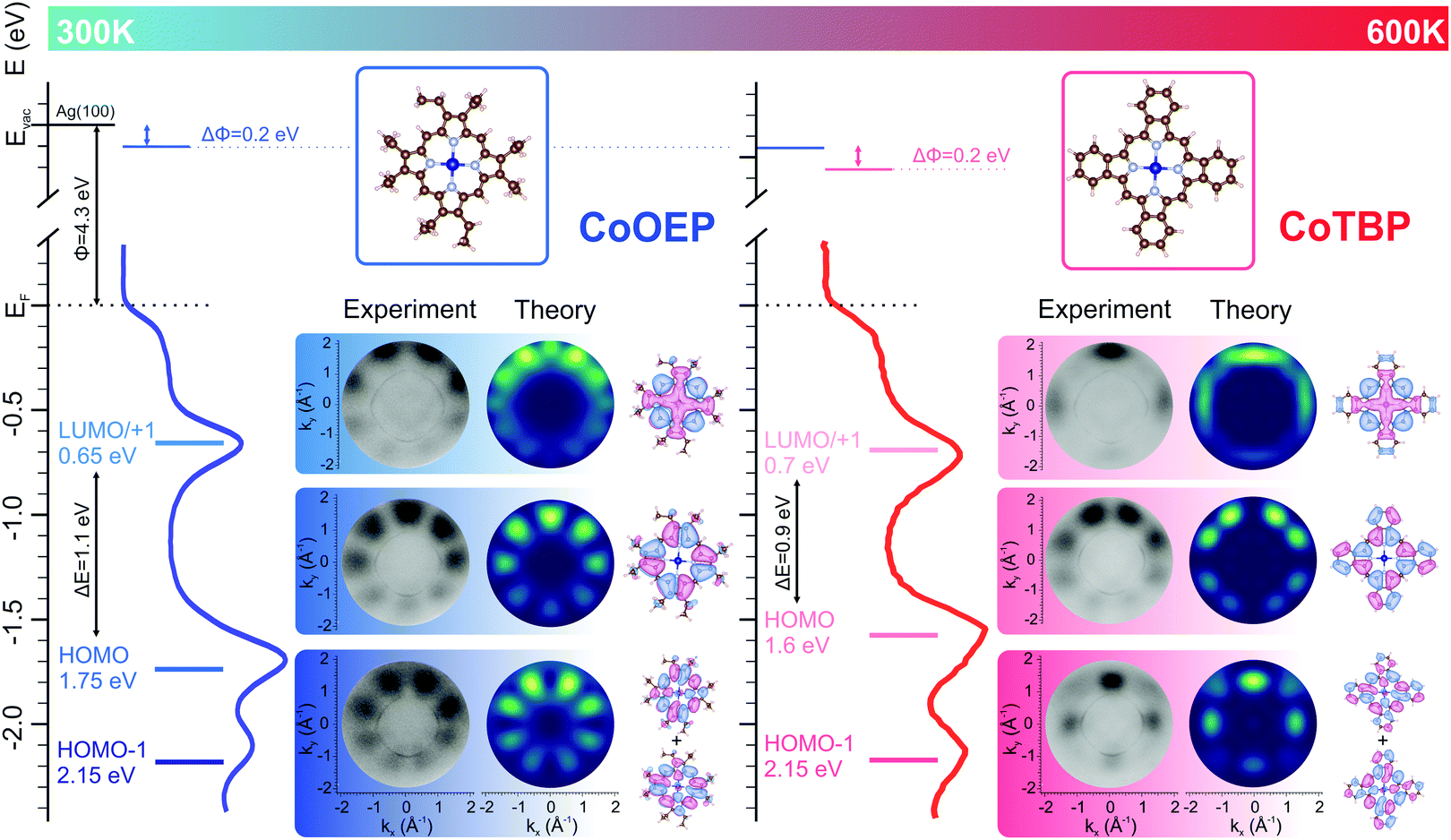 | ||
| Fig. 1 Angle-integrated UPS spectra and schematic energy diagrams of electronic levels of the CoOEP/Ag(100) and CoTBP/Ag(100) interfaces. Molecular orbitals of CoOEP and CoTBP, and corresponding experimental and theoretical momentum maps at the indicated BE, are shown. | ||
On silver, the M-OEP interaction with the surface leads to an electron charge transfer from the metal substrate to the molecule.1,9 As a consequence, in the case of CoOEP deposited on silver, near-edge X-ray absorption fine structure (NEXAFS) and ultraviolet photoemission spectroscopy (UPS) suggested the hybridization between the Co d-states and the Ag sp-band, resulting in a modification of the valence states of the adsorbed molecule with respect to the free OEP molecule.9,10 In particular, the new state appearing at 0.65 eV below the Fermi level, in the monolayer phase of CoTPP on Ag(110),10 CoOEP on Ag(111),9 and FeTPP on Ag(111),11 was unanimously assigned to the filling of the metal dz2 unoccupied molecular orbital.
In the past, the photoemission tomography (PT) approach12 has been employed to study the geometry and the electronic structure of the macrocycle in self-assembled porphyrin and its derivatives.13–15 Additionally, it has been applied to study the pathway of thermally-activated on-surface intermolecular reactions.16 In the present work, we will show that the PT approach can also be extended to the study of surface-induced intramolecular modifications, i.e., ring-closure reaction, in single molecules self-assembled in molecular arrays. More frequently, intramolecular reactions are monitored by STM, temperature-programmed desorption (TPD), X-ray photoemission spectroscopy (XPS) and NEXAFS techniques,2–6,17,18 which do not always allow the authoritative identification of an adsorbed species. Only indirect conclusions can be made from, e.g., the presence or absence of specific vibrational modes in infrared spectroscopy or chemical shifts in core-level spectroscopy. Moreover, although providing an excellent insight into the molecular backbone structure, STM is less conclusive about the molecular periphery, where adsorbates tend to react with the substrate, and the geometrical and chemical changes observed are convoluted with the substrate electron density changes,15 leaving room for alternative interpretations. The kinetics of the ring-closure reaction depends on a fine interplay between steric hindrance among adjacent molecules and substrate reactivity.
The first factor is influenced by the degree of molecular ordering and surface density: the tighter the molecular packing the lower the ring-closure reaction rate (higher temperature required). The interaction with the substrate governs two rate limiting processes: (i) the on-surface recombination of hydrogen (gasH2), and (ii) the residence time of the dynamically fluctuating peripheral terminations in the favourable (co-planar) transient geometry. The latter process depends on the strength of interaction of porphyrin terminations with the substrate, which is mostly governed by van der Waals forces, and hence can be very large for aromatic groups on poorly reactive metals (2H-TPP cyclo-dehydrogenation starting at 545 K on copper19), and very small on highly reactive substrates such as oxides (the same at 650–700 K on titanium dioxide6).
Here we will follow the possible on-surface chemical modification of CoOEP molecules assembled in a densely packed monolayer array on Ag(100) by annealing up to 600 K. The annealing treatment may result in the transformation from CoOEP to CoTBP via the complete dehydrogenation of the ethyl groups in the pristine porphyrin molecules, followed by the formation of six-membered carbon rings. The imaging of molecular orbitals in the reciprocal space, being extremely sensitive to the chemical structure, allows us to directly probe the ring-closure reaction (CoOEP → CoTBP) on the Ag(100) surface. We unequivocally determined the electronic occupancy and energy level alignment of the molecular frontier orbitals, i.e., the highest occupied (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of the as-deposited CoOEP array and the CoTBP ad-layer formed by the on-surface reaction. Moreover, the reaction has been confirmed by XPS and NEXAFS measured at the N K-edge. For both CoOEP and CoTBP molecular assemblies, the charge transfer at the organic/metal interface induces the reduction of the cobalt ion in the metal-organic arrays, as verified by Co 2p core-level and Co L3-edge NEXAFS measurements.
In Fig. 1 we present the angle-integrated valence band spectra of pristine and annealed CoOEP/Ag(100) interfaces. While the valence band spectrum of the bare Ag(100) substrate has a rather featureless plateau associated with the sp-bands (see Fig. S1, ESI†), three prominent features can be recognized for the organic/metal interfaces, both before and after annealing. In particular, the feature peaked at higher binding energy (BE = 2.15 eV) is present for both the pristine and annealed samples, while the two low energy peaks undergo only minor changes in the energy position. While those energy shifts cannot reveal the nature of the chemical modifications, clear differences are observed in the measured momentum maps. To identify the origin of the features observed in the UPS spectra and test the possible CoOEP → CoTBP on-surface reaction, the experimental maps have been compared to the square modulus of the Fourier transform (FT) of the real space molecular orbitals, calculated for gas-phase CoOEP and CoTBP molecules. Indeed, it has been shown for various organic/metal interfaces that there exists a one-to-one correspondence between the momentum distribution of the photocurrent and the molecular orbitals in the reciprocal space.12–15,20 In that way, the features observed for the CoOEP/Ag(100) system can be unequivocally assigned to the molecular states simulated for the gas-phase CoOEP, where the states centered at BE 2.15 eV, 1.75 eV and 0.65 eV are due to the HOMO−1, HOMO and degenerate LUMO/LUMO+1, respectively. No match for the annealed interface can be found between the measured maps and the CoOEP calculated maps, while the momentum maps simulated for CoTBP match very well the three measured patterns at 2.15 eV, 1.60 eV and 0.70 eV for the annealed interface, which according to the present calculation can be assigned to the HOMO−1, HOMO and LUMO/LUMO+1 molecular states of CoTBP, respectively. The excellent agreement between the measured and simulated maps using the PT approach is direct proof that the annealing at 600K on Ag(100) induces the chemical modification of CoOEP and leads to the formation of the tetrabenzoporphyrin array.
For both systems, the HOMO–LUMO gaps are significantly lowered after on surface deposition, from 3.0 eV to 1.1 eV (CoOEP) and from 2.5 eV to 0.9 eV (CoTBP), suggesting a strong molecule-surface interaction at both interfaces. Furthermore, the presence of the LUMOs for both the organic systems confirms the charge transfer effect for both metalorganic/Ag(100) systems. The occupation of the former π* gas-phase LUMOs, delocalized over the entire molecular macrocycle, has been previously observed for metal porphyrin and phthalocyanine molecules on metal surfaces.13,15 After proving the CoOEP → CoTBP on-surface reaction by PT, in the next we shed light on the energy level alignment of CoOEP and CoTBP molecules assembled on Ag(100).
The adsorbate-induced work function (WF) changes in CoOEP and CoTBP are extracted from the shift of the secondary electron cut-offs. All changes are referred to the value for the bare Ag(100) work function (4.3 eV). Upon the adsorption of CoOEP, the WF of the metal substrate decreases by 0.2 eV, and it further decreases to 3.9 eV after the formation of CoTBP. With the entire set of experimental data, we can now draw the energy level alignment picture for CoOEP and CoTBP molecular arrays on Ag(100) substrate (see Fig. 1). Next, it is interesting to reveal the possible contribution of the atomic like dz2 orbital to the lowest-in-energy peak (BE = 0.65 eV) in the valence band of CoOEP/Ag(100), stated in previous studies.9,10 To this end, we analyse the DFT simulations showing that it is the molecular orbital LUMO+2 that has a strong atomic like dz2 character. Following the same procedure as for the molecular patterns reported in Fig. 1, we simulated the map for LUMO+2, shown in Fig. S3 (ESI†). Clearly, the present simulated pattern does not resemble the corresponding experimental map measured at BE 0.65 eV reported in Fig. 1, and, therefore, we can exclude the main contribution from the dz2 state (LUMO+2) to the corresponding valence band feature, contrary to the expectation from previous reports for Co and Fe porphyrins on silver.9–11
The changes observed in the N K-edge NEXAFS (Fig. 2a) and C 1s XPS spectra (Fig. S2, ESI†) acquired before and after the annealing of the CoOEP/Ag(100) interface to 600 K fully support the CoOEP → CoTBP on-surface transformation (see the ESI† for details).
 | ||
| Fig. 2 N K-edge (a), Co 2p3/2 (b) and Co L3-edge spectra (c) of CoOEP and CoTBP on Ag(100). | ||
The ring-closure reaction in the porphyrin array can also alter the oxidation state of the Co ion in the adsorbed CoOEP and CoTBP molecules and, as a consequence, modify the magnetic properties in the organic array. To investigate the Co oxidation state, we performed XPS measurements of the Co 2p3/2 core-level, together with NEXAFS experiments at the Co L3-edge.
In Fig. 2b, we compare the Co 2p3/2 core-level spectra of CoOEP and CoTBP adsorbed on the Ag(100) substrate. The XPS spectrum of CoOEP shows a main line peaked at 778.8 eV and a broad satellite at higher BE (with the maximum at 780.9 eV). Compared to the CoOEP multilayer phase reported in ref. 9, we observe a significant shift of the Co 2p3/2 signal (−1.8 eV), as well as a notable change in the satellite features. This finding can be attributed to the reduction of the metal ion in the CoOEP monolayer phase9,21 caused by strong charge transfer at the organic/metal interface, as observed in the UPS measurements (Fig. 1). After annealing, the Co 2p spectrum of CoTBP shows the same spectral shape of the pristine layer, but a small energy shift (∼0.2 eV) of the main peak to higher BE. This shift may stem from the charge redistribution from the tetra-pyrrolic pocket to the more extended aromatic macrocycle of CoTBP that weakens the metal-ligand field, hence reducing the Co to N charge transfer. We remark that such a shift results from the combination with additional contributions in the opposite direction, such as the aforementioned decrease of the work function, and a possible increase of surface screening of the core-hole due to closer surface proximity of CoTBP. To get direct access to the oxidation state of the Co ion, we acquired the Co L3-edge absorption spectra for both CoOEP and CoTBP (see Fig. 2c). And compared the results with the published NEXAFS and X-ray Magnetic Circular Dichroism (XMCD) measurements reported by Arruda et al.4 for CoOEP and CoTBP on copper. The NEXAFS spectra in Fig. 2c show a similar shape and energy position of the resonances for both CoOEP and CoTBP, suggesting a similarity in the electronic structure, as well as in the oxidation and spin states of the Co ion in the two different metalorganic arrays. The NEXAFS spectra measured with s- and p-polarized light across the Co L3-edge display a significant difference in the energy position of the resonances. While in the spectra measured in s-polarisation where the main feature is peaked at 779.5 eV (peak A), the spectra recorded with p-polarization show a peak at lower photon energy (778.7 eV, peak B). Both spectra show a pronounced shoulder (peak C) at higher photon energies (see Fig. 2b). The observed linear dichroism for the low energy feature in the Co L3-edge spectra points to an out-of-plane component, which can be associated with the dz2 orbital.4 The full occupation of the dz2 orbital would be reflected in the experimental data by the absence of the low energy feature in the absorption spectra. Notably, the complete filling of this orbital in similar molecular compounds on copper surfaces was responsible for the quenching of the spin moment, as evident from XMCD data.4 In contrast, the Co L3-edge spectra of CoOEP and CoTBP on Ag(100) are similar to the published spectra of CoOEP on Cu(100).4 Following the XMCD measurements and theoretical calculations reported in ref. 4, and the XPS and NEXAFS measurements shown here, we propose that the charge transfer at the organic/metal interface stabilizes the cobalt ion in CoOEP and CoTBP on Ag(100) in a non-integer valence state, with a formal oxidation state between +1 and +2.
In conclusion, we show that PT is an efficient tool to probe on-surface reactions in self-assembled molecular arrays. This has been proved through the temperature-induced CoOEP → CoTBP transformation on Ag(100), which goes through the dehydrogenation of two neighbouring ethyl groups in CoOEP followed by the formation of a six-membered carbon ring, and the method can be extended to the study of different π-conjugated molecules on various conductive substrates. While PT can be an appropriate method to study on surface complete dehydrogenation reactions in a molecular array, STM is still a more suitable approach to follow the degree of dehydrogenation at lower temperatures where different molecular intermediate products are still present.
Combining different surface science techniques, accompanied by DFT calculations, we also show that the electronic structure and the energy alignment in both CoOEP and CoTBP arrays are very similar. In particular, the strong interaction of both organic molecules with the silver substrate at the interface causes charge transfer from the substrate to the molecule, as clearly identified by the PT, resulting in the occupation of the gas phase LUMO/+1 delocalized over the entire molecular macrocycle. Furthermore, the strong interaction at the metal/organic interface is also responsible for the reduction of the cobalt ion in both CoOEP and CoTBP to an oxidation state between +1 and +2, as found from Co L3-edge NEXAFS measurements.
P.P. acknowledges the support from the Austrian Science Fund (FWF) project I3731. The computational results presented have been achieved in part using the Vienna Scientific Cluster (VSC).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259 CrossRef CAS.
- J. Xiao, S. Ditze, M. Chen, F. Buchner, M. Stark, M. Drost, H.-P. Steinrück, J. M. Gottfried and H. Marbach, J. Phys. Chem. C, 2012, 116, 12275 CrossRef CAS.
- G. Di Santo, S. Blankenburg, C. Castellarin-Cudia, M. Fanetti, P. Borghetti, L. Sangaletti, L. Floreano, A. Verdini, E. Magnano, F. Bondino, C. A. Pignedoli, M.-T. Nguyen, R. Gaspari, D. Passerone and A. Goldoni, Chem. – Eur. J., 2011, 17, 14354 CrossRef.
- L. M. Arruda, M. E. Ali, M. Bernien, N. Hatter, F. Nickel, L. Kipgen, C. F. Hermanns, T. Bißwanger, P. Loche, B. Heinrich, K. J. Franke, P. M. Oppeneer and W. Kuch, Phys. Chem. Chem. Phys., 2020, 22, 12688 RSC.
- L. M. Arruda, M. E. Ali, M. Bernien, F. Nickel, J. Kopprasch, C. Czekelius, P. M. Oppeneer and W. Kuch, J. Phys. Chem. C, 2019, 123, 14547 CrossRef CAS.
- G. Lovat, D. Forrer, M. Abadia, M. Dominguez, M. Casarin, C. Rogero, A. Vittadini and L. Floreano, Nanoscale, 2017, 9, 11694 RSC.
- B. W. Heinrich, G. Ahmadi, V. L. Müller, L. Braun, J. I. Pascual and K. J. Franke, Nano Lett., 2013, 13, 4840 CrossRef CAS.
- D. van Vörden, M. Lange, M. Schmuck, J. Schaffert, M. C. Cottin, C. A. Bobisch and R. Möller, J. Chem. Phys., 2013, 138, 211102 CrossRef.
- Y. Bai, F. Buchner, I. Kellner, M. Schmid, F. Vollnhals, H.-P. Steinrück, H. Marbach and J. M. Gottfried, New J. Phys., 2009, 11, 125004 CrossRef.
- M. Fanetti, A. Calzolari, P. Vilmercati, C. Castellarin-Cudia, P. Borghetti, G. Di Santo, L. Floreano, A. Verdini, A. Cossaro, I. Vobornik, E. Annese, F. Bondino, S. Fabris and A. Goldoni, J. Phys. Chem. C, 2011, 115, 11560 CrossRef CAS.
- G. Di Santo, C. Sfiligoj, C. Castellarin-Cudia, A. Verdini, A. Cossaro, A. Morgante, L. Floreano and A. Goldoni, Chem. – Eur. J., 2012, 18, 12619 CrossRef CAS.
- P. Puschnig, S. Berkebile, A. J. Fleming, G. Koller, K. Emtsev, T. Seyller, J. D. Riley, C. Ambrosch-Draxl, F. P. Netzer and M. G. Ramsey, Science, 2009, 326, 702 CrossRef CAS.
- G. Zamborlini, D. Lüftner, Z. Feng, B. Kollmann, P. Puschnig, C. Dri, M. Panighel, G. Di Santo, A. Goldoni, G. Comelli, M. Jugovac, V. Feyer and C. M. Schneider, Nat. Commun., 2017, 8, 1 CrossRef.
- P. Kliuiev, G. Zamborlini, M. Jugovac, Y. Gurdal, K. von Arx, K. Waltar, S. Schnidrig, R. Alberto, M. Iannuzzi, V. Feyer, M. Hengsberger, J. Osterwalder and L. Castiglioni, Nat. Commun., 2019, 10, 5255 CrossRef.
- H. M. Sturmeit, I. Cojocariu, M. Jugovac, A. Cossaro, A. Verdini, L. Floreano, A. Sala, G. Comelli, S. Moro, M. Stredansky, M. Corva, E. Vesselli, P. Puschnig, C. M. Schneider, V. Feyer, G. Zamborlini and M. Cinchetti, J. Mater. Chem. C, 2020, 8, 8876 RSC.
- X. Yang, L. Egger, P. Hurdax, H. Kaser, D. Lüftner, F. C. Bocquet, G. Koller, A. Gottwald, P. Tegeder, M. Richter, M. G. Ramsey, P. Puschnig and S. Soubatch, Nat. Commun., 2019, 10, 3189 CrossRef.
- M. Röckert, M. Franke, Q. Tariq, S. Ditze, M. W. Stark, P. Uffinger, D. Wechsler, U. Singh, J. Xiao, H. Marbach, H. P. Steinrück and O. Lytken, Chem. – Eur. J., 2014, 20, 8948 CrossRef.
- I. Cojocariu, H. M. Sturmeit, G. Zamborlini, A. Cossaro, A. Verdini, L. Floreano, E. D’Incecco, M. Stredansky, E. Vesselli, M. Jugovac, M. Cinchetti, V. Feyer and C. M. Schneider, Appl. Surf. Sci., 2020, 504, 144343 CrossRef CAS.
- M. M. Franke, Q. Tariq, D. Lungerich, N. Jux, M. Stark, A. Kaftan, S. Ditze, H. Marbach, M. Laurin, J. Libuda, H.-P. Steinrück and O. Lytken, J. Phys. Chem. C, 2014, 118, 26729 CrossRef.
- M. Wießner, D. Hauschild, C. Sauer, V. Feyer, A. Schöll and F. Reinert, Nat. Commun., 2014, 5, 4156 CrossRef.
- G. Zamborlini, M. Jugovac, A. Cossaro, A. Verdini, L. Floreano, D. Lüftner, P. Puschnig, V. Feyer and C. M. Schneider, Chem. Commun., 2018, 54, 13423 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc00311a |
| This journal is © The Royal Society of Chemistry 2021 |