
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResorcin[4]arene-based multidentate phosphate ligands with superior binding affinity for nanocrystal surfaces†
Suren J.
Nemat
 a,
Dietger
Van den Eynden
a,
Loren
Deblock
ab,
Michael
Heilmann
a,
Jesper M.
Köster
a,
Mahsa
Parvizian
a,
Konrad
Tiefenbacher
*ac and
Jonathan
De Roo
*a
a,
Dietger
Van den Eynden
a,
Loren
Deblock
ab,
Michael
Heilmann
a,
Jesper M.
Köster
a,
Mahsa
Parvizian
a,
Konrad
Tiefenbacher
*ac and
Jonathan
De Roo
*a
aDepartment of Chemistry, University of Basel, Basel 4058, Switzerland. E-mail: Konrad.Tiefenbacher@unibas.ch; Jonathan.DeRoo@unibas.ch
bDepartment of Chemistry, Ghent University, Gent, 9000, Belgium
cDepartment of Biosystems Science and Engineering, ETH Zürich, Basel, CH-4058, Switzerland
First published on 13th April 2021
Abstract
We designed and synthesized two resorcin[4]arene scaffolds with four phosphate binding groups. The ligands effectively bind in at least a tridentate fashion at low surface coverage. The superior binding affinity is demonstrated using solution NMR spectroscopy and exceeds that of single phosphonates.
Colloidal nanocrystals have a high surface-to-volume ratio, hence their surface chemistry is equally important to their function as the nanocrystal core itself.1,2 A colloidal nanocrystal is often a hybrid object, with organic surfactants (ligands) adsorbed on an inorganic core. Over the last decade, researchers have uncovered the specific binding modes of nanocrystal–ligand interactions,3–6 and determined the rules for maximizing the nanocrystal solubility.7–10 For a given nanocrystal surface, ligands can be ranked according to their binding strength. For CdSe and InP nanocrystals, thiolates and phosphonates are excellent ligands while carboxylates are average and amines/phosphines are poor binders.11–14 Thiols are also excellent ligands for gold, but have low affinity for metal oxides.15 Carboxylates bind well to metal oxides but phosphonates are superior ligands.16 One way of increasing the binding affinity is multidenticity; i.e., adding multiple binding groups on a single scaffold. For carboxylates, this has been realized with derivatives of citric acid or EDTA.17,18 Thiolates can be upgraded to carbodithioate,19 dithiocarbamate,20 or dithiolate.21 The Mattoussi group developed polymeric ligands with catechol, imidazole, thiol, and phosphonic acid groups.22
In an effort to design a high-affinity ligand, combining the atomic precision of small molecules and the high binding affinity of multidentate ligands, we previously developed anthracene 1,8 diphosphoric acid derivatives (Fig. 1).23 These ligands quantitatively displace oleate ligands from CdSe nanocrystals in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry, confirming the bidentate binding. While they are useful in nanocrystal-sensitized upconversion,23 they are also light-sensitive, and the rigid anthracene backbone precludes multidenticity (>2) and limits the conformational flexibility of the phosphoric acid moieties, leading to suboptimal coordination with surface metal ions. Therefore, we synthesized and investigated here two new ligands 1 and 2 based on the resorcinarene scaffold (Fig. 1). Ligand 1 contains a resorcin[4]arene24,25 macrocycle comprised of four phosphate substituted aromatic units. The macrocycle shows some degree of flexibility, as the structure rapidly interconverts between two equivalent boat conformations, but full rotation is prevented through its alkyl feet (Fig. S18–S20, ESI†). Resorcin[4]arene 2 was designed to have even higher conformational flexibility. It contains four highly flexible terminal phosphates on its alkyl feet, while its upper rim alkyl chain modifications ensure comparable size and molecular weight of ligands 1 and 2 (Table S2 and Fig. S28, ESI†).
:2 stoichiometry, confirming the bidentate binding. While they are useful in nanocrystal-sensitized upconversion,23 they are also light-sensitive, and the rigid anthracene backbone precludes multidenticity (>2) and limits the conformational flexibility of the phosphoric acid moieties, leading to suboptimal coordination with surface metal ions. Therefore, we synthesized and investigated here two new ligands 1 and 2 based on the resorcinarene scaffold (Fig. 1). Ligand 1 contains a resorcin[4]arene24,25 macrocycle comprised of four phosphate substituted aromatic units. The macrocycle shows some degree of flexibility, as the structure rapidly interconverts between two equivalent boat conformations, but full rotation is prevented through its alkyl feet (Fig. S18–S20, ESI†). Resorcin[4]arene 2 was designed to have even higher conformational flexibility. It contains four highly flexible terminal phosphates on its alkyl feet, while its upper rim alkyl chain modifications ensure comparable size and molecular weight of ligands 1 and 2 (Table S2 and Fig. S28, ESI†).
 | ||
| Fig. 1 Chemical structure and key properties of the previously synthesized bidentate phosphate ligand and the macrocyclic multidentate phosphate ligands developed in this work. | ||
Macrocyclic ligands have been considered for nanocrystals before, but past research was mostly focused on noble metal nanocrystals, featuring soft ligands.26–29 In addition, their binding to the nanocrystal surface was not thoroughly studied. Therefore, 1 and 2 present the first systematic effort in macrocyclic ligand design for a wider class of nanocrystals. Here we take ZrO2 nanocrystals as a model system to evaluate the binding affinity of 1 and 2 in detail through an X-for-X type exchange.3 The possibility for Z-type displacement make CdSe nanocrystals less useful as model system.4
The route towards compound 1 starts from the readily available tetramethoxy resorcin[4]arene 3,30 which has proven to be a versatile starting material for tetra-functionalized, C4-symmetric resorcin[4]arenes (Scheme 1a).31,32 After phosphorylation to the protected phosphate 4 and subsequent deprotection using TMSBr, compound 1 was obtained in 58% yield over two steps. The reaction progress of the deprotection was closely tracked by NMR-spectroscopy to achieve complete conversion of 4 with minimal formation of side products. Ligand 2 was obtained in a three step synthesis starting from hydroxyl-footed resorcin[4]arene 5 (Scheme 1b).33 The upper rim of 5 was alkylated to produce 6, which was converted to the phenyl phosphate 7. The switch to phenyl protecting groups in this synthetic sequence was essential as ethyl protecting groups would not be cleaved selectively in the presence of the alkylphosphate of compound 2. Hydrogenolysis using platinum oxide in the presence of acetic acid effectively removed the phenyl protection groups of 7 to yield ligand 2 in 45% yield over three steps. The optimization of the deprotection conditions proved to be the crucial step of this synthetic sequence. Employing Pd/C as the heterogeneous catalyst leads to the formation of side products, while running the hydrogenolysis with platinum oxide in absence of acetic acid resulted in incomplete conversion of 7. Ligands 1 and 2 were thus obtained from commercially available material in only three and four steps, respectively. These synthetic routes compare favourably to the synthesis of the bidentate anthracene ligand (5 steps).23 The tetraphosphate resorcin[4]arenes 1 and 2 are bench and solution stable chemicals (see ESI† chapter 3.5), which were fully characterized by ESI-HRMS, NMR- and IR spectroscopy (see ESI†).
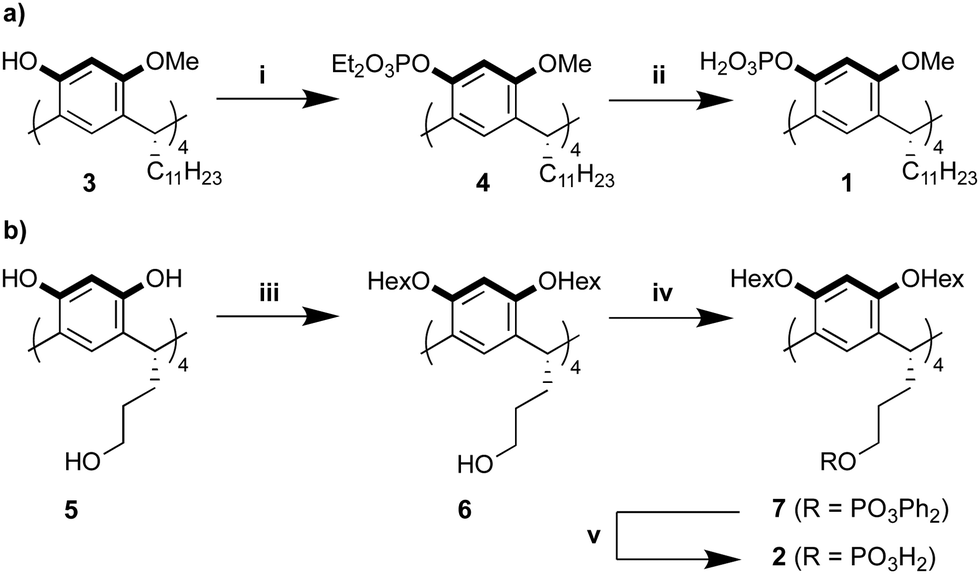 | ||
| Scheme 1 (a) Synthesis of ligand 1. (b) Synthesis of ligand 2. Reagents and conditions: (i) diethyl chlorophosphate, NaH, THF, 0 °C to r.t., 16 h, 67%; (ii) TMSBr, DCM, −78 °C to r.t., 48 h, then MeOH, r.t., 1 h, 86%; (iii) 1-iodohexane, K2CO3, acetone, r.t. to 70 °C, 7 d, 60%; (iv) diphenyl chlorophosphate, NEt3, DCM, 0 °C to r.t., 16 h, 82%; (v) H2, PtO2, acetic acid, EtOH, r.t., 24 h, 91%. | ||
To gain detailed insight in the affinity of 1 and 2 toward nanocrystal surfaces, we used oleate capped ZrO2 nanocrystals as model system. The nanocrystals were synthesized from ZrCl4 and benzyl alcohol, according to an established procedure and are about 5 nm in size.34 The nanocrystals were purified by precipitation/redispersion cycles (using acetone/chloroform) until a monolayer of oleate is present on the surface, as attested by the broad resonances in the 1H NMR spectrum in CDCl3 (Fig. S1, ESI†). Ligands bound to nanocrystals feature both homogeneous and heterogeneous line broadening in NMR,35 and therefore, the line width is typically used as an indicator for ligand binding. Unfortunately, the ligands 1 and 2 are poorly soluble in CDCl3 and therefore we were forced to analyse the competitive binding in THF-d8. In THF, a small part of the oleate ligand auto-desorbs as oleic acid,36 evidenced by a small sharp feature on top of the broadened resonances (Fig. S2, ESI†). This is common in polar and coordinating solvents, like THF.4,37 The amount of auto-desorption varied from 3–10% depending also on the water content of the sample. When we titrated 1 into a suspension of nanocrystals in THF-d8, we saw a gradual increase in the sharp resonances of oleic acid, indicating further displacement of oleate from the surface, see Fig. 2.
 | ||
| Fig. 2 (A) Scheme of the ligand exchange. This is a cartoon and not meant to indicate a specific surface configuration. (B) Waterfall plot of the 1H NMR spectra of the titration of 1 into a suspension of oleate capped ZrO2 nanocrystals (16.7 mg mL−1), ([oleic acid] = 14 mM). The 1H NMR spectra of pure nanocrystals and pure 1 are also shown. The resonance with the * label belongs to a pool of exchangeable protons; an average of water and the acidic protons of 1. | ||
In Fig. 2, we show the region of the alkene resonance of oleate/oleic acid (5.4 ppm) since there is no spectral overlap of this alkene resonance with ligand 1. The full-range spectra and additional titration steps are shown in Fig. S3 (ESI†). From Fig. 2, we observe that almost all oleate was removed from the surface upon addition of 0.36 equivalents of 1. Note that one equivalent of 1 corresponds to 4 equivalents of phosphate with respect to oleic acid. The titration with 2 showed similar results and is shown in Fig. S4 (ESI†). The fraction of bound oleate was quantified and plotted in Fig. 3A. Until about 0.15 equivalents, the bound fraction decreases linearly with a slope of −3.3 ± 0.2 for ligand 1 and a slope of −3.2 ± 0.1 for ligand 2. Within error, both ligands have a similar binding affinity and at least three of the four phosphate groups on the macrocycle displace a carboxylate functionality from the nanocrystal surface. Therefore, they bind at least in a tridentate fashion and possibly even truly tetradentate at the start of the titration. This is however obscured by the small auto-desorption of oleic acid in THF. Finally, upon adding more than 0.15 equivalents, the slope decreases, presumably due to the effect of steric crowding (see below) and only around the addition of 0.4 equivalents, all oleate is removed from the ZrO2 surface.
 | ||
| Fig. 3 (A) Decrease of bound oleic acid ligands over de course of the titration with 1 or 2. Linear fits to the initial decrease are also plotted. (B) Lattice model where a binding site is a square, oleate is a blue, striped circle and ligand 1 or 2 are represented by the three connected orange circles. | ||
The varying slope of the displacement can be easily understood with a simple lattice model (Fig. 3B). We represent each binding site as a square, the oleate as a blue, striped circle and the macrocycle as three connected orange circles, (occupying three adjacent squares). At low equivalents of the macrocycle, each new macrocycle ligand can bind free from interference with the other macrocycles. However, at about 0.2 equivalents of macrocycle, the surface mole fraction χ has increased significantly to 0.33 and the fractional surface occupied by the macrocycle has increased to 60%. The remaining 40% surface sites are most likely randomly distributed over the surface. From the lattice model in Fig. 3B, it is clear that the probability of finding three free adjacent squares has dropped dramatically. A kinetically impeded surface reorganization needs to occur to accommodate another tridentate ligand, or the macrocycle is forced to bind in bi- or mono-dentate fashion. Our data supports the latter hypothesis. In addition, the similar binding isotherms of 1 and 2 indicate that both have enough conformational flexibility to find the optimal configuration on the nanocrystal surface. The limiting factor is finding three (or four) free binding sites that are adjacent.
After addition of about one equivalent of macrocycle, all originally bound oleate is displaced from the surface. This is not only evidenced by the narrow line width of the alkene resonance in the 1H NMR spectrum (Fig. 2) but also by the absence of the alkene resonance in the diffusion filtered spectrum (Fig. S6, ESI†). A diffusion filtered spectrum only retains signals from nanocrystal bound ligands.35 The nanocrystals were purified by precipitation with acetone and redispersed in THF-d8 (see SI). The resulting 1H NMR spectrum is dominated by broad CH2 and CH3 signals belonging to the alkyl chains of 1 (C11H23), indicating a successful purification and removal of all oleate (Fig. S6, ESI†). As expected, the resonances of 1 which are close to the surface are extremely broadened, and disappear in the background. The inorganic core retained its monoclinic crystal structure during the ligand exchange according to XRD measurements (Fig. S7, ESI†).
In the 31P NMR spectrum, an asymmetric peak shape is observed for bound 1 around −5 ppm (Fig. S8, ESI†). In contrast, a symmetric phosphate peak is observed for bound 2 (Fig. 4). The width of the resonance is approximately 2000 Hz, consistent with previous reports on bound phosphates and phosphonates.23,38 To further test the binding strength of 2, we added hexylphosphonic acid38 to the purified nanocrystals. Upon addition of 2 or 4 equivalents, some bound hexylphosphonate is observed as a broad feature around 25 ppm but no change to the resonance of 2 is detected. This is probably due to free binding sites that are being occupied by hexylphosphonate. There is a large amount of hexylphosphonic acid that remains free (sharp resonance), which is not displacing 2 from the surface. Similar results are obtained in competition experiments with oleylphosphonic acid for both ligand 1 and ligand 2 (Fig. S8, ESI†). Finally, the purified nanocrystals are not aggregated in solution and remain colloidally stable (Fig. S9, ESI†).
 | ||
| Fig. 4 31P NMR spectrum of a purified ZrO2 nanocrystal suspension, functionalized by 2, and after addition of 2 or 4 equivalents hexylphosphonic acid. | ||
In conclusion, we presented the concise synthesis of two bench stable macrocyclic ligands with phosphate binding groups that behave effectively like (at least) tridentate ligands towards ZrO2 nanocrystal surfaces. Even phosphonic acids cannot displace the resorcin[4]arene ligands and thus these ligands are among the strongest binders available, if not the strongest small molecule ligands (excluding polymers). However, also a limitation of multidentate ligands is presented; they only bind according to their maximum denticity at low surface coverage.
The authors thank the SNSF NCCR Molecular Systems Engineering (project number: 182895) and FWO Flanders for funding. The support of Dr Michael Pfeffer concerning HR-MS analysis, and Prof. Daniel Häussinger concerning VT-NMR experiments is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119(8), 4819–4880 CrossRef CAS PubMed.
- J. De Roo, K. De Keukeleere, Z. Hens and I. V. Driessche, Dalton Trans., 2016, 45, 13277–13283 RSC.
- N. C. Anderson, M. P. Hendricks, J. J. Choi and J. S. Owen, J. Am. Chem. Soc., 2013, 135, 18536–18548 CrossRef CAS PubMed.
- Y. Yao and W. E. Buhro, Chem. Mater., 2020, 32, 205–214 CrossRef CAS.
- J. Zhang, H. Zhang, W. Cao, Z. Pang, J. Li, Y. Shu, C. Zhu, X. Kong, L. Wang and X. Peng, J. Am. Chem. Soc., 2019, 141, 15675–15683 CrossRef CAS PubMed.
- Y. Yang, H. Qin, M. Jiang, L. Lin, T. Fu, X. Dai, Z. Zhang, Y. Niu, H. Cao, Y. Jin, F. Zhao and X. Peng, Nano Lett., 2016, 16, 2133–2138 CrossRef CAS PubMed.
- Y. Yang, H. Qin and X. Peng, Nano Lett., 2016, 16, 2127–2132 CrossRef CAS PubMed.
- T. Kister, D. Monego, P. Mulvaney, A. Widmer-Cooper and T. Kraus, ACS Nano, 2018, 12, 5969–5977 CrossRef CAS PubMed.
- D. Monego, T. Kister, N. Kirkwood, P. Mulvaney, A. Widmer-Cooper and T. Kraus, Langmuir, 2018, 34, 12982–12989 CrossRef CAS PubMed.
- N. C. Anderson, P. E. Chen, A. K. Buckley, J. De Roo and J. S. Owen, J. Am. Chem. Soc., 2018, 140, 7199–7205 CrossRef CAS PubMed.
- R. R. Knauf, J. C. Lennox and J. L. Dempsey, Chem. Mater., 2016, 28, 4762–4770 CrossRef CAS.
- R. Gomes, A. Hassinen, A. Szczygiel, Q. A. Zhao, A. Vantomme, J. C. Martins and Z. Hens, J. Phys. Chem. Lett., 2011, 2, 145–152 CrossRef CAS.
- A. Ritchhart and B. M. Cossairt, Inorg. Chem., 2019, 58(4), 2840–2847 CrossRef CAS PubMed.
- X. Liu, M. Yu, H. Kim, M. Mameli and F. Stellacci, Nat. Commun., 2012, 3, 1182 CrossRef PubMed.
- K. De Keukeleere, S. Coucke, E. De Canck, P. Van Der Voort, F. Delpech, Y. Coppel, Z. Hens, I. Van Driessche, J. S. Owen and J. D. Roo, Chem. Mater., 2017, 29, 10233–10242 CrossRef CAS.
- M. Hellstern, M. Gantenbein, L. Le Pleux, G. Puebla-Hellmann, E. Lörtscher and M. Mayor, Adv. Mater. Interfaces, 2019, 6 Search PubMed.
- L. M. Bishop, J. C. Yeager, X. Chen, J. N. Wheeler, M. D. Torelli, M. C. Benson, S. D. Burke, J. A. Pedersen and R. J. Hamers, Langmuir, 2012, 28, 1322–1329 CrossRef CAS PubMed.
- C. Querner, P. Reiss, J. Bleuse and A. Pron, J. Am. Chem. Soc., 2004, 126, 11574–11582 CrossRef CAS PubMed.
- M. T. Frederick and E. A. Weiss, ACS Nano, 2010, 4, 3195–3200 CrossRef CAS PubMed.
- N. Zhan, G. Palui, A. Kapur, V. Palomo, P. E. Dawson and H. Mattoussi, J. Am. Chem. Soc., 2015, 137, 16084–16097 CrossRef CAS PubMed.
- W. Wang and H. Mattoussi, Acc. Chem. Res., 2020, 53(6), 1124–1138 CrossRef CAS PubMed.
- J. De Roo, Z. Huang, N. J. Schuster, L. S. Hamachi, D. N. Congreve, Z. Xu, P. Xia, D. A. Fishman, T. Lian, J. S. Owen and M. L. Tang, Chem. Mater., 2020, 32, 1461–1466 CrossRef CAS.
- A. G. S. Hoegberg, J. Am. Chem. Soc., 1980, 102, 6046–6050 CrossRef CAS.
- L. M. Tunstad, J. A. Tucker, E. Dalcanale, J. Weiser, J. A. Bryant, J. C. Sherman, R. C. Helgeson, C. B. Knobler and D. J. Cram, The, J. Org. Chem., 1989, 54, 1305–1312 CrossRef CAS.
- C. Schottle, E. L. Clark, A. Harker, A. Solovyov, A. T. Bell and A. Katz, Chem. Commun., 2017, 53, 10870–10873 RSC.
- J.-M. Ha, A. Solovyov and A. Katz, J. Phys. Chem. C, 2010, 114, 16060–16070 CrossRef CAS.
- G. Vyas, S. Bhatt and P. Paul, ACS Omega, 2019, 4, 3860–3870 CrossRef CAS PubMed.
- J. Hassinen, P. Pulkkinen, E. Kalenius, T. Pradeep, H. Tenhu, H. Häkkinen and R. H. A. Ras, J. Phys. Chem. Lett., 2014, 5, 585–589 CrossRef CAS PubMed.
- M. J. McIldowie, M. Mocerino, B. W. Skelton and A. H. White, Org. Lett., 2000, 2, 3869–3871 CrossRef CAS PubMed.
- S. J. Nemat, H. Jędrzejewska, A. Prescimone, A. Szumna and K. Tiefenbacher, Org. Lett., 2020, 22, 5506–5510 CrossRef CAS PubMed.
- J. N. Smith and N. T. Lucas, Chem. Commun., 2018, 54, 4716–4719 RSC.
- B. C. Gibb, R. G. Chapman and J. C. Sherman, J. Org. Chem., 1996, 61, 1505–1509 CrossRef CAS.
- J. De Roo, F. Van den Broeck, K. De Keukeleere, J. C. Martins, I. Van Driessche and Z. Hens, J. Am. Chem. Soc., 2014, 136, 9650–9657 CrossRef CAS PubMed.
- J. De Roo, N. Yazdani, E. Drijvers, A. Lauria, J. Maes, J. S. Owen, I. Van Driessche, M. Niederberger, V. Wood, J. C. Martins, I. Infante and Z. Hens, Chem. Mater., 2018, 30, 5485–5492 CrossRef CAS.
- J. D. Roo, Y. Justo, K. D. Keukeleere, F. Van denBroeck, J. C. Martins, I. V. Driessche and Z. Hens, Angew. Chem., Int. Ed., 2015, 54, 6488–6491 CrossRef PubMed.
- R. Grisorio, D. Debellis, G. P. Suranna, G. Gigli and C. Giansante, Angew. Chem., Int. Ed., 2016, 55, 6628–6633 CrossRef CAS PubMed.
- J. De Roo, Z. Zhou, J. Wang, L. Deblock, A. J. Crosby, J. S. Owen and S. S. Nonnenmann, Chem. Mater., 2018, 30, 8034–8039 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc00223f |
| This journal is © The Royal Society of Chemistry 2021 |