
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co-self-assembly of multiple DNA origami nanostructures in a single pot†
Joshua A.
Johnson
a,
Vasiliki
Kolliopoulos
b and
Carlos E.
Castro
 *ac
*ac
aBiophysics Graduate Program, The Ohio State University, 281 W Lane Ave, Columbus, OH 43210, USA. E-mail: castro.39@osu.edu
bDepartment of Chemical and Biomolecular Engineering, The Ohio State University, 281 W Lane Ave, Columbus, OH 43210, USA
cDepartment of Mechanical and Aerospace Engineering, The Ohio State University, 281 W Lane Ave, Columbus, OH 43210, USA
First published on 15th April 2021
Abstract
Simultaneous self-assembly of two distinct DNA origami nanostructures folded with the same scaffold strand was achieved in a single pot. Relative yields were tuned by adjusting concentrations of the competing strands, correlating well with folding kinetics of individual structures. These results can faciliate efficient fabrication of multi-structure systems and materials.
Structural DNA nanotechnology1 is a powerful approach for the self-assembly of structures with precisely programmed geometry and function. DNA origami,2–4 in particular, enables the design of nanodevices with complex shape, tunable mechanical properties, and programmable dynamics.5–7 In this approach, a mixture of many synthetic ssDNA strands, referred to as staple strands, bind to two or more distant segments of a long circular strand of ssDNA, referred to as the scaffold, which is typically 7000–8000 nucleotides long. Staple binding orchestrates folding of the scaffold strand into precisely defined shapes with nanometer or even sub-nanometer precision8 with potential applications in biosensing,9–11 drug delivery,12–14 and nanofabrication.15–17 While these devices have demonstrated tremendous promise, the details of self-assembly are not fully understood. A better understanding is critical to improving rapid and high yield assembly and enabling efficient workflows to design application-specific devices including complex systems consisting of multiple structures.
Previous studies have revealed the primary factors driving DNA origami self-assembly are the entropy cost associated with forming scaffold loops as staples bring two different domains together18–20 and the kinetics of binding staples which serve as nucleators early in the self-assembly process.21,22 A critical parameter within these studies is whether or not the folding reactions occurred in thermal equilibrium (typically by slow annealing from 90–95 °C23) or out of equilibrium by jumping to a critical temperature as in the case of isothermal folding protocols,24 by chemical quenching,25,26 or by mechanical means.27 In the case of slow annealing, the most thermodynamically stable local regions of an origami fold first, enhancing cooperativity of binding for some nearby regions. Interestingly, if there are multiple sets of staples, each coding for a different nanostructure, domains of different structures can fold within the same scaffold to create a chimera.28 However, in studies with more rapid changes to folding conditions, staples with faster binding kinetics drive nucleation, and folding the remainder of a structure proceeds very quickly. Yields of complete structures remain high despite the greater likelihood of falling into kinetic traps.21,24,27
Here we demonstrate that in a mixture of staple sets for multiple structures, tuning the kinetics of folding in out-of-equilibrium folding protocols, we achieve complete assembly of multiple distinct structures. We show it is possible to effectively bifurcate the folding pathway to co-self-assemble two different user-defined rod-like structures, 6-helix bundle (6HB) and 18-helix bundle (18HB) DNA origami nanostructures (Fig. 1A and B), simultaneously in one pot with tuneable ratios. We first characterized folding of the individual structures at various staple strand concentrations and isothermal annealing temperatures. Our results reveal that the kinetics of self-assembly for structures folded separately correlate with yields during co-self-assembly and indicate that early folding events are key in regulating mixed folding results.
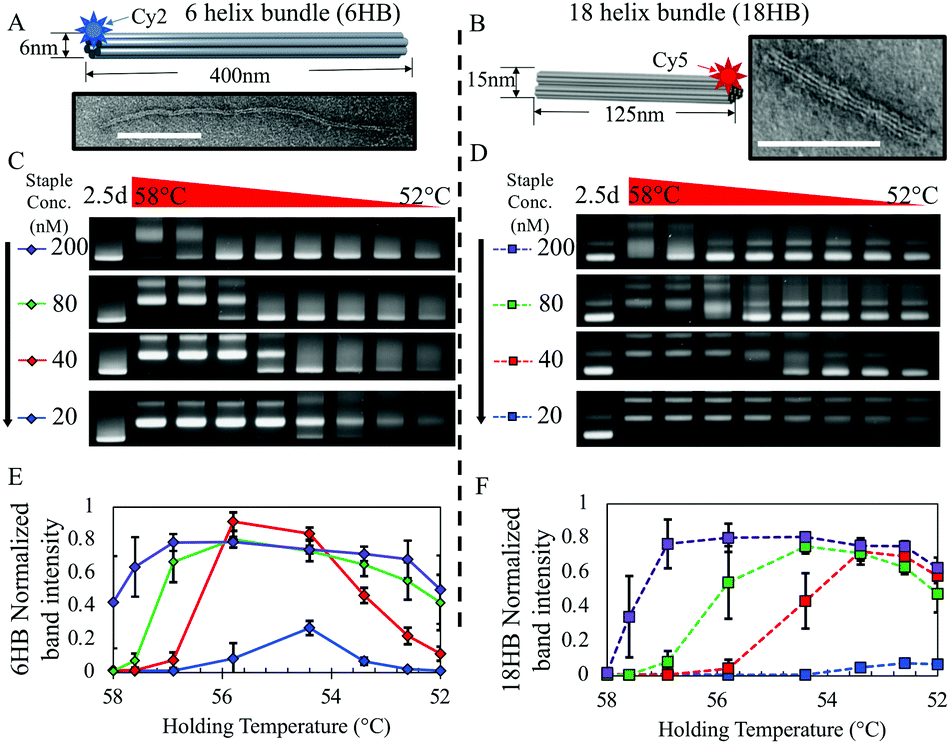 | ||
| Fig. 1 (A and B) Solid models of 6HB and 18HB structures with corresponding TEM images of individually folded structures. Scale bars are 100 nm. (C and D) Gel shift assay of isothermal annealing protocol for 6HB and 18HB structures at 200 nM, 80 nM, 40 nM, 20 nM staple concentrations (lane 1 shows a 2.5 day annealing protocol, and lanes 2 through 9 show folding constant annealing temperatures at 58, 57.6, 56.9, 55.8, 54.4, 53.4, 52.6, and 52 °C). Full gel images and corresponding fluorescence gel scans are provided (Fig. S3–S11, ESI†). (E and F) Plots show corresponding band intensity for 6HB and 18HB gel results quantified from triplicate data. Intensities were normalized against the 2.5 day annealing control. | ||
For each assembly reaction, we followed previously established protocols for isothermal folding24 testing constant annealing temperatures initially in the range of 40–60 °C (Fig. S1, ESI†) then narrowed our range to 52–58 °C (Fig. 1C and D). At high staple concentrations, both structures exhibited high yield folding over most of the temperature range, while the range of temperatures at which structures fold clearly varies between the 6HB and 18HB for lower staple concentrations.
In particular, at 40 nM staple concentration there is a ∼2 °C decrease in the upper bound of temperatures at which the 6HB folds while the 18HB exhibits a more dramatic decrease in the upper bound. Additionally, the maximum yield for both structures appears to be nearly insensitive to staple concentrations over the range of 10-fold down to 2-fold excess; however, higher staple concentrations are more robust against temperature variations (i.e. larger range of high yield annealing temperatures). Reducing the staple strand concentrations to be equimolar with the scaffold concentration (20 nM) results in a large reduction in yield. These results suggest staple strands can be conserved without sacrificing folding yield for these structures, at least down to 2-fold excess, but at the cost of requiring more careful consideration and control of the annealing temperature. Most importantly, the range of temperatures and concentrations at which these two structures fold are distinct from one another, especially at lower staple concentrations, which suggests that in a mixture containing both staple sets we could find regimes where one structure or the other folds preferentially. At 53 °C we see a significant variability in yields between the 6HB and 18HB while both still exhibit a reasonable yield of folding. Thus, we chose to use 53 °C isothermal folding reactions for all subsequent experiments.
We characterized the folding kinetics for both structures by quenching folding reactions in liquid nitrogen at specific timepoints during isothermal annealing to evaluate folding progression by gel electrophoresis. We found that at early annealing times, a band corresponding to partially folded structures appeared with a slower mobility than well folded structures. We quantified the intensities of gel bands corresponding to both partially folded and well folded structures and fit these data to a simple 3-state model.
In this model, we simplified the kinetics of DNA origami folding to an initial unfolded state, an intermediate partially folded state, and a completely folded state. We only consider the forward reaction rates and assume that the reverse reaction rates are negligible giving only two rate parameters (details in ESI†). The chemical reaction is depicted below:
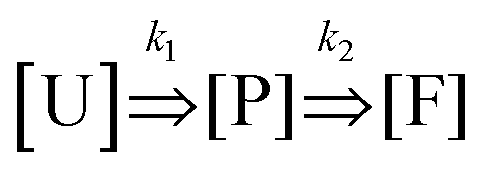 | (1) |
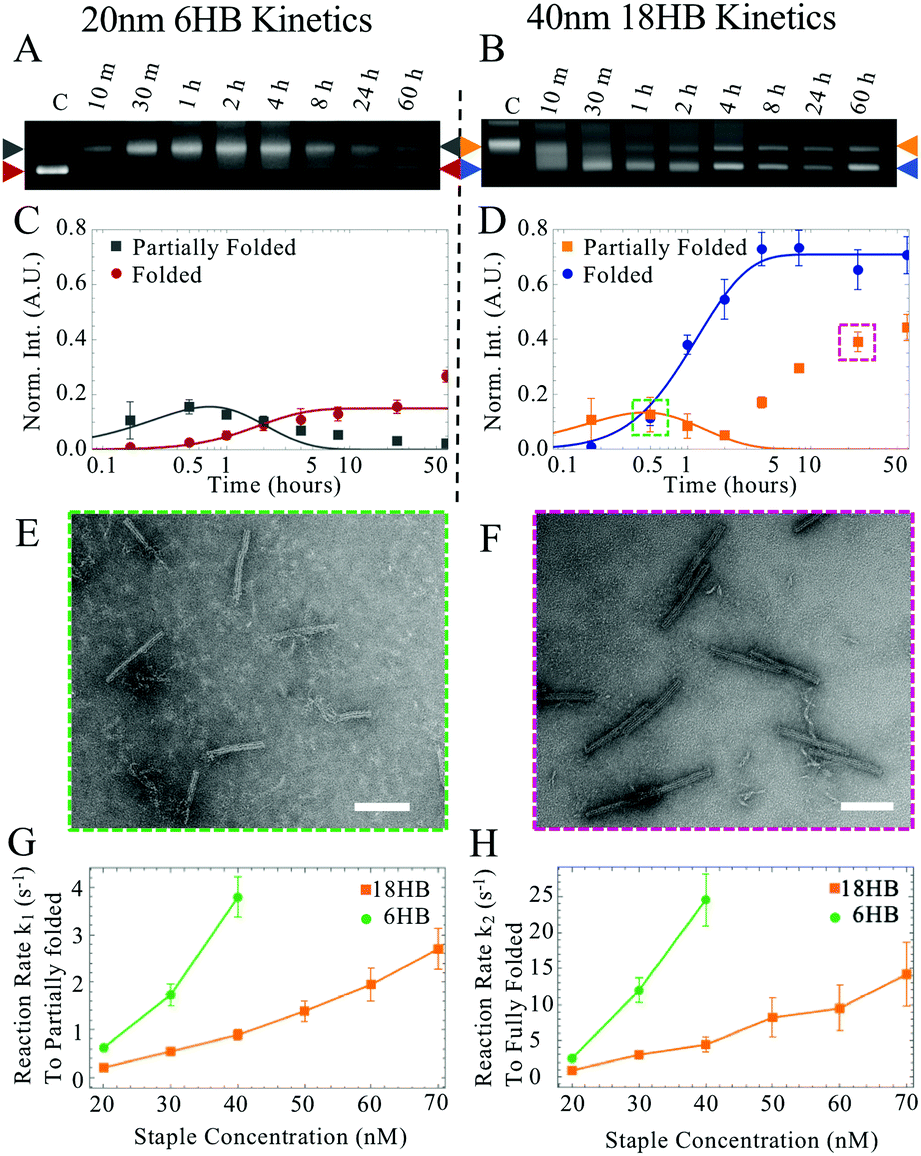 | ||
| Fig. 2 Example of comparison of kinetics for (A) 6HB origami folded with 20 nM staple concentration and (B) 18HB origami folded with 40 nM staple concentration. Quantification of kinetics from gel data for 6HB (C) and 18HB (D). TEM images of structures excised from lagging bands in 40 nM 18HB kinetics at 30 min (E) and 24 h (F) showing partially folded and dimer structures, respectively. Scale bars are 100 nm. Effective rate constants k1 (G) and k2 (H) for 20–40 nM 6HB staple strand concentrations (green) and 20–70 nM 18HB staple strand concentrations (orange). | ||
At later time points for the 18HB, a narrower peak corresponding to dimer structures appears (Fig. 2B, D and H) which has a gel mobility similar to partially folded structures at earlier time points. To avoid considering these dimers, we weighted earlier time points more heavily in fitting than later time points (Fig. S2–S11, ESI†). Examples of folding kinetics results for the 6HB and the 18HB are shown in Fig. 2A–H. A summary of all extracted rates is provided in Fig. 2(G and H). These results demonstrate kinetics of folding vary strongly with staple concentration and higher concentrations of 18HB staples are required to match the kinetics of folding for 6HB.
Over this range of concentrations, the 6HB folding kinetics are faster than the 18HB, even for conditions where the overall yields are lower. We hypothesize that the cooperative nature of folding combined with isothermal annealing protocols will drive co-self-assembly in a manner which favors structures with faster folding kinetics; hence, at similar staple concentrations the 6HB would more likely fold to completion over the slower folding 18HB. The fitted folding rates for the individual 6HB and 18HB suggest that the 18HB would need approximately 1.5 to 2 times greater concentration of staples to achieve similar folding rates to the 6HB (Table S1, ESI†). This suggests the 18HB may outcompete the 6HB in co-self-assembly when the 18HB staples are at higher concentrations. To test our hypothesis, we characterized co-self-assembly reactions involving a range of both 6HB and 18HB staples to determine if any structures fold to completion and whether the relative yields of structures are consistent with predictions based on the kinetics of individual structures. Our results show that mostly well-folded 6HB appear in the folding reactions with both staple sets at standard concentrations (200 nM) (Fig. S12, ESI†). This is a subtle but critical result demonstrating that a DNA origami nanostructure can successfully fold to completion in a mixture of interfering strands. Furthermore, the result that the 6HB outcompetes the 18HB is consistent with the general trend of the 6HB exhibiting faster folding kinetics at similar staple concentrations. We proceeded with co-self-assembly reactions using concentration ranges over which the folding kinetics of each structure were comparable. We used a fixed amount of 6HB at low concentrations (20, 30, and 40 nM) and a larger range of 18HB concentrations from 0 nM to 120 nM (Fig. 3). Each structure included an overhang to bind a fluorescently labeled oligo to allow quantification of folding. From laser-scanned agarose gels and TEM images, we found that low concentrations of 18HB staples resulted in well-formed 6HB. Intermediate 18HB concentrations resulted in a mixture of both well-formed 18HB and 6HB, while high 18HB concentrations resulted in well-formed 18HB structures. For the case of 20 nM 6HB staples, both structures successfully self-assemble at roughly equal ratios when the 18HB staples were also at ∼20 nM. The cases with 30 nM and 40 nM 6HB staples require ∼1.5–2× more 18HB staples to achieve equal amounts of folded structures. Individual color channels for scanned gels and additional TEM images are provided in Fig. S13–S15 (ESI†). With this data, we can map out conditions which primarily produce 18HB, 6HB, or a desired ratio of both. Therefore, without altering staple designs, it is possible to bifurcate the 6HB and 18HB folding pathways to co-self-assemble two distinct populations in desired quantities by varying staple concentrations in a single-pot reaction. Closer examination of the yields of each structure in competitive folding reactions revealed that low concentrations of 18HB staples, in fact, increased yields of 6HB structures as shown in Fig. 2c. This result is somewhat unexpected considering typical behaviors of biomolecules in competitive binding assays and seems to indicate some amount of competitor facilitated folding.
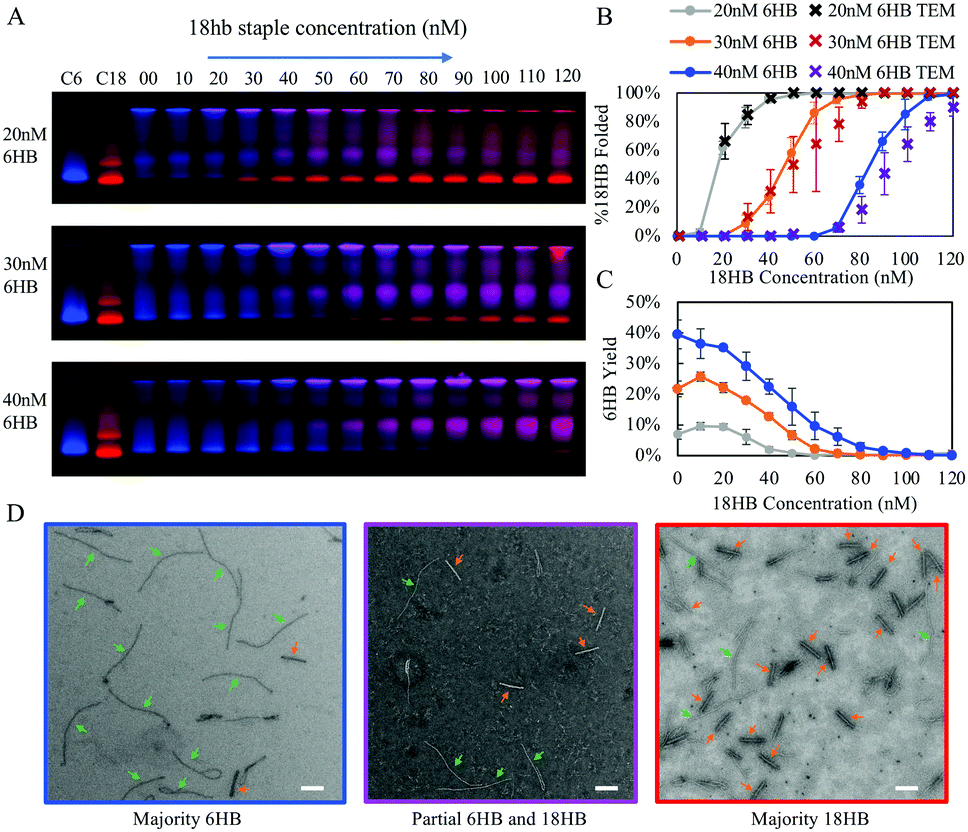 | ||
| Fig. 3 (A) Gel shift assay of 6HB staples kept constant at 40 nM, 30 nM, or 20 nM with titrations of 18HB staples from 0 nM to 120 nM in increments of 10 nM showing a transition from 6HB folded structures to 18HB folded structures. Lanes C6 and C18 are controls with structures folded at standard 200 nM staple concentrations. (B) Quantification of gels run in triplicate with presented as the fraction of band intensity corresponding to 18HB divided by total band intensity of regions containing both 6HB and 18HB. (C) Band intestines of 6HB normalized to control showing competitor facilitated folding of 6HB at lower concentrations. (D) Confirmation via TEM show 6HB structures and 18HB from the same folding reaction from excised gel bands. Images are representative of samples folded with 18HB staple concentrations around their respective transition from mostly 6HB structures to mostly 18HB structures but the particular conditions for chosen images are all with 40 nM 6HB staples and 70 nM (left), 90 nM (middle), and 110 nM (right) 18HB staples. Scale bars are 100 nm. | ||
We have shown that folding reactions containing a mixture of staple strands from two distinct DNA origami nanostructures, a 6HB and 18HB, can be tuned to result in fully folded versions of each in a single pot reaction. We found that a greater fraction of 6HB folded compared to 18HB unless 18HB staples were ∼1.5–2 fold the concentration of 6HB staples. These results correlate with our characterization of folding kinetics in which a 1.5–2 fold greater concentration of 18HB staples was necessary to match the folding rates of 6HB, even in folding conditions where 18HB exhibited higher yields. Since the folding rates of 6HB are faster, the early formation of partially folded regions of 6HB allow it to dominate co-self-assembly reactions. These partially folded regions of 6HB likely organize the remaining scaffold in a configuration that inhibits 18HB formation. Specifically, since the 6HB is rather long, upon partial folding some regions of scaffold may become physically separated enough to prohibit the formation of scaffold loops necessary for 18HB nucleation (i.e. piecewise binding of 18HB staples). TEM imaging of partially folded states in combined folding reactions (Fig. S16, ESI†) suggest sections of 6HB form first, which supports the idea that kinetics of early folding events are important and that these partially folded structures may topologically inhibit binding of staples from the 18HB. This illustrates how the topology of nucleating domains likely influences folding pathways. Structural frustration at nucleation sites has previously been shown to impact folding for DNA origami with different isomers.29 Our results suggest these factors can also impact the formation of entirely different structures. Also, a critical difference between the 6HB and 18HB is in packing of helices in the bundle. All bundles in the 6HB structure have only 2 neighboring helices, resulting in fewer crossovers per bundle. Thus, 6HB staples are more likely to have longer binding regions than the 18HB whose core helices necessarily have 3 neighboring helices. The longer binding domains of 6HB would both thermodynamically and kinetically favor 6HB formation (Section S1.7, ESI†). A closer look at the staple strand designs reveals a region of the 6HB with many staples that have segments with relatively high melting temperatures (Fig. S39, ESI†), which may also play a role in favoring 6HB folding.
Another interesting phenomenon observed in our data is the increased yields of 6HB formation with low concentrations of 18HB staples. We might expect that the introduction of 18HB staples facilitates rearrangement or restructuring of misfolded structures. Since the 6HB exhibits greater yields in individual folding reactions at slightly higher temperatures, introduction of competing DNA may offer a similar benefit to increased temperature. It is possible that some 18HB strands, which happen to occupy some of the scaffold effectively increase the ratio of 6HBs to available scaffold. Additionally, 18HB strands that bind to two distant regions of the scaffold could effectively pay the entropic penalty to facilitate binding of 6HB strands.
Further exploration into the details of DNA origami self-assembly using co-self-assembly reactions could reveal design principles for directing assembly through topologically controlled pathways.30 Our approach could be combined with existing higher-order assembly schemes to create more complex single-pot structures through multi-component hierarchical assembly. This would be especially useful for creating higher-order structures in complex environments such as within hydrogels or in biological environments where delivery of a fully assembled construct is challenging. Combining co-self-assembly reactions with DNA-based computing or sensing mechanisms, this work opens the possibility for smart nanofabrication where the type of nanostructure formed can be programmed as a function of environmental conditions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. C. Seeman, J. Theor. Biol., 1982, 35, 237–247 CrossRef.
- P. K. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- C. E. Castro, F. Kilchherr, D. N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221 CrossRef CAS PubMed.
- A. E. Marras, L. Zhou, H. J. Su and C. E. Castro, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 713–718 CrossRef CAS PubMed.
- C. E. Castro, H. J. Su, A. E. Marras, L. Zhou and J. Johnson, Nanoscale, 2015, 7, 5913–5921 RSC.
- F. Wang, X. Zhang, X. Liu, C. Fan and Q. Li, Small, 2019, 15, 1900013 CrossRef PubMed.
- J. J. Funke and H. Dietz, Nat. Nanotechnol., 2016, 11, 47–52 CrossRef CAS PubMed.
- A. Puchkova, C. Vietz, E. Pibiri, B. Wünsch, M. Sanz Paz, G. P. Acuna and P. Tinnefeld, Nano Lett., 2015, 15, 8354–8359 CrossRef CAS PubMed.
- Y. Huang, M. K. Nguyen, A. K. Natarajan, V. H. Nguyen and A. Kuzyk, ACS Appl. Mater Interfaces, 2018, 51, 44221–44225 CrossRef PubMed.
- M. W. Hudoba, Y. Luo, A. Zacharieas, M. G. Poirier and C. E. Castro, ACS Nano, 2017, 11, 6566–6573 CrossRef CAS PubMed.
- P. D. Halley, C. R. Lucas, E. M. McWilliams, M. J. Weber, R. A. Patton, C. Kural, D. M. Lucas, J. C. Byrd and C. E. Castro, Small, 2016, 12, 308–320 CrossRef CAS PubMed.
- S. Li, Q. Jiang, S. Liu, Y. Zhang, Y. Tian, C. Song, J. Wang, Y. Zou, G. J. Anderson, J. Y. Han and Y. Chang, Nat. Biotechnol., 2018, 36, 258 CrossRef CAS PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed.
- G. Tikhomirov, P. Peterson and L. Qian, Nature, 2017, 552, 67–71 CrossRef CAS PubMed.
- X. Liu, F. Zhang, X. Jang, M. Pan, P. Liu, W. Li, B. Zhu, J. Li, H. Chen, L. Wang and J. Lin, Nature, 2018, 559, 593–598 CrossRef CAS PubMed.
- B. Uprety, J. Jensen, B. R. Aryal, R. C. Davis, A. T. Woolley and J. N. Harb, Langmuir, 2017, 33, 10143–10152 CrossRef CAS PubMed.
- K. E. Dunn, F. Dannenberg, T. E. Ouldridge, M. KwiatKowska, A. J. Turberfield and J. Bath, Nature, 2015, 525, 82–86 CrossRef CAS PubMed.
- J. Lee Tin Wah, C. David, S. Rudiuk, D. Baigl and A. Estévez-Torres, ACS Nano, 2016, 10, 1978–1987 CrossRef CAS PubMed.
- J. M. Majikes, P. N. Patrone, D. Schiffels, M. Zwolak, A. J. Kearsley, S. P. Forry and J. A. Liddle, Nucleic Acids Res., 2020, 48, 5268–5280 CrossRef CAS PubMed.
- F. Schnieder, N. Möritz and H. Dietz, Sci. Adv., 2019, 5, eaaw1412 CrossRef PubMed.
- F. Dannenberg, K. E. Dunn, J. Bath, M. Kwaitkowska, A. J. Turberfield and T. E. Ouldridge, J. Chem. Phys., 2015, 143, 10B617 CrossRef PubMed.
- M. Sujfutdinow, W. M. Jacobs, A. Reinhardt, C. Schneider and D. M. Smith, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5877–E5886 CrossRef PubMed.
- J. P. J. Sobczak, T. G. Martin, T. Gerling and H. Dietz, Science, 2012, 338, 1458–1461 CrossRef CAS PubMed.
- J. M. Majikes, J. A. Nash and T. H. LaBean, Nanoscale, 2017, 9, 1637–1644 RSC.
- B. Högberg, T. Liedl and W. M. Shih, J. Am. Chem. Soc., 2009, 131, 9154–9155 CrossRef PubMed.
- W. Bae, K. Kim, D. Min, J. K. Ryu, C. Hyeon and T. Y. Yoon, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- J. M. Majikes, J. A. Nash and T. H. LaBean, New J. Phys., 2016, 18, 115001 CrossRef.
- R. Kosinski, A. Mukhortava, W. Pfeifer, A. Candelli, P. Rauch and B. Saccà, Nat. Commun., 2019, 10, 1–12 CrossRef CAS PubMed.
- A. E. Marras, L. Zhou, V. Kolliopoulos, H. J. Su and C. E. Castro, New J. Phys., 2016, 18, 055005 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of material and methods, kinetics characterization and modelling, and additional TEM images. See DOI: 10.1039/d1cc00049g |
| This journal is © The Royal Society of Chemistry 2021 |