
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsolable small-molecule cysteine sulfenic acid†
Tsukasa
Sano
,
Ryosuke
Masuda
,
Shohei
Sase
and
Kei
Goto
 *
*
Department of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, 152-8551, Japan. E-mail: goto@chem.titech.ac.jp
First published on 15th February 2021
Abstract
An isolable small-molecule cysteine sulfenic acid (Cys–SOH) protected by a molecular cradle was synthesized by direct oxidation of the corresponding cysteine thiol and its structure was established by X-ray crystallographic analysis. Studies on biologically relevant reactivity indicated its usefulness as a biorepresentative small-molecule sulfenic acid model.
Sulfenic acids (R–SOH) are well recognized as important intermediates in biological1,2 and organic3 transformations. Cysteine sulfenic acids (Cys–SOH), in particular, have been attracting increasing attention due to their crucial roles in a variety of cellular processes, including transcription regulation, signal transduction, and the regulation of oxygen metabolism and oxidative stress responses.1 Oxidation of cysteine thiols (Cys–SH) to Cys–SOH by reactive oxygen species is a reversible post-translational modification, and the Cys–SOH formed can then be converted to the disulfides (Cys–SSR) and to the cysteine sulfinic acids (Cys–SO2H). Although much research has been carried out into their biological functions, Cys–SOH still remains an elusive species due to the intrinsic instability of the R–SOH species. A Cys–SOH readily undergoes dehydrative self-condensation to produce a thiosulfinate (R–S(O)–S–R).3a Cys–SOH generated in several proteins are stabilized by the local protein environment, and their existence has been supported by analytical methods such as X-ray crystallography, mass spectrometry, and NMR spectroscopy.4 In most cases, however, identification of Cys–SOH in protein has been based on indirect methods, i.e., trapping experiments utilizing chemical probes.5 For the modeling of protein Cys–SOH, many studies on small-molecule Cys–SOH have also been carried out; however, the greater instability of small-molecule Cys–SOH compared with those stabilized in proteins has been a significant hurdle in efforts to determine their chemical properties and reactivity. There are no reports of even spectroscopic observations of small-molecule Cys–SOH. In the field of organosulfur chemistry, various non-cysteinyl sulfenic acids have been synthesized by taking advantage of either kinetic stabilization or thermodynamic stabilization.6–8 The chemical behavior of Cys–SOH has been interpreted based on the properties and reactivities of those non-cysteinyl sulfenic acids although their structural and electronic properties are different from those of sulfenic acids with a cysteine backbone. For a better understanding of the chemical behavior of Cys–SOH in biological systems, the development of a stable reference compound for Cys–SOH is highly desirable.
Here we report the first synthesis and isolation of a stable small-molecule Cys–SOH utilizing a nanosized molecular cavity as a protective cradle for the reactive cysteine unit (Fig. 1). Crystallographic analysis of the cradled Cys–SOH and model studies of biologically relevant chemical processes involving Cys–SOH are also described.
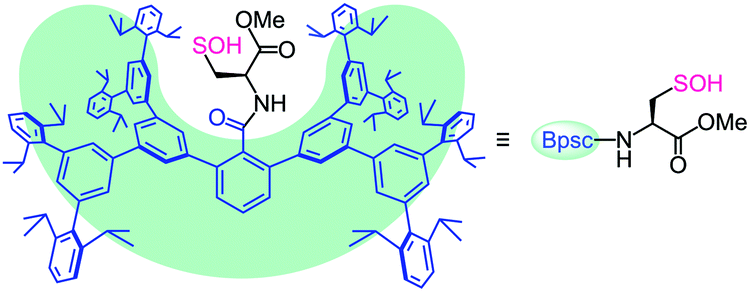 | ||
| Fig. 1 Cradled cysteine model for stabilization of cysteine-derived reactive intermediates such as Cys–SOH. | ||
The bulky groups that have been utilized for kinetic stabilization of sulfenic acids6,7 have only a local protective effect limited to the directly attached reactive functionality and are therefore too ineffective in stabilizing a whole cysteine unit with the –SOH functionality. During the course of our studies on highly reactive species of biological importance,9,10 we recently designed a molecular cavity based on an m-phenylene dendrimer framework of ca. 2 nm scale.9d In the present study, we utilized the framework as a molecular cradle that can accommodate a whole cysteine unit within the cavity (Fig. 1). In this model, a cavity-shaped benzoyl group (denoted as Bpsc) is introduced to a cysteine unit as an N-terminal protecting group. This novel cysteine model, which is referred to as a cradled cysteine, is expected to be useful for the synthesis of reference compounds of highly reactive cysteine derivatives such as Cys–SOH.
Carboxylic acid 1 (Bpsc–OH) with the m-phenylene dendrimer framework was synthesized by the route shown in Scheme S1 (see ESI†). After conversion of 1 to acid chloride 2, the cradle moiety was introduced to a cysteine unit by the reaction of 2 with the cysteine derivative 3 to afford 4 (Scheme 1). The reduction of 4 with dithiothreitol (DTT) afforded the cradled Cys–SH 5 in 91% yield.
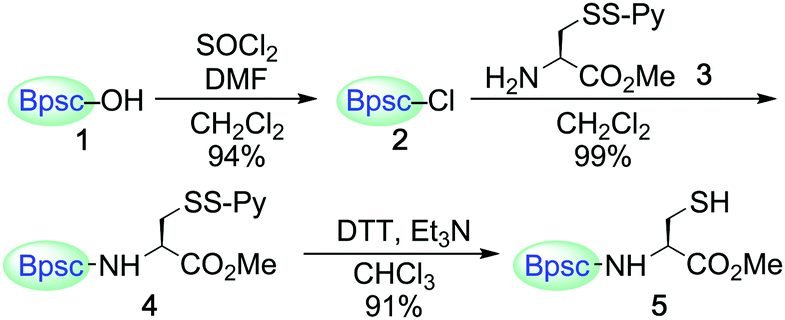 | ||
| Scheme 1 Synthesis of Cys–SH 5. Py represents 2-pyridyl. | ||
The reaction of Cys–SH with H2O2 is central to many processes in biology, from protein folding to redox signaling.11 The initial products have been commonly assumed to be Cys–SOH, but there is no example in which this has been directly demonstrated, besides in the crystallographic monitoring of protein thiol oxidation.4c,d For non-cysteinyl derivatives, the spectroscopic monitoring of the formation of 9-fluorotriptycenesulfenic acid by H2O2 oxidation of the corresponding thiol was recently reported.12 Herein, the synthesis of an isolable Cys–SOH by direct H2O2 oxidation of Cys–SH was examined. Treatment of Cys–SH 5 with aqueous sodium hydroxide followed by the reaction with H2O2 in THF/H2O (ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at room temperature afforded the corresponding Cys–SOH 6 as the major product; it was isolated as stable colorless crystals in 27% yield (Scheme 2). An organic base could also be used; in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), H2O2 oxidation of 5 in THF afforded 6 in an isolated yield of 36%. This is the first example of the synthesis and isolation of a small-molecule Cys–SOH. It is notable that 6 can be purified by silica gel column chromatography in air and handled under standard laboratory conditions. Characterization of 6 was performed by NMR and IR spectroscopies, as well as by elemental analysis. The 1H NMR (C6D6) spectrum of 6 exhibited a singlet due to the SOH proton at δ 5.36, which is readily exchangeable with D2O.
:1 v/v) at room temperature afforded the corresponding Cys–SOH 6 as the major product; it was isolated as stable colorless crystals in 27% yield (Scheme 2). An organic base could also be used; in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), H2O2 oxidation of 5 in THF afforded 6 in an isolated yield of 36%. This is the first example of the synthesis and isolation of a small-molecule Cys–SOH. It is notable that 6 can be purified by silica gel column chromatography in air and handled under standard laboratory conditions. Characterization of 6 was performed by NMR and IR spectroscopies, as well as by elemental analysis. The 1H NMR (C6D6) spectrum of 6 exhibited a singlet due to the SOH proton at δ 5.36, which is readily exchangeable with D2O.
 | ||
| Scheme 2 Synthesis of Cys–SOH 6 by H2O2 oxidation of Cys–SH 5. | ||
The structure of 6 was finally established by X-ray crystallographic analysis (Fig. 2). The cysteine moiety of 6 is anchored in a large cavity produced by the Bpsc cradle (dimensions ca. 2.3 × 1.7 nm). The interatomic distance between sulfur and oxygen of the SOH moiety of 6 (1.616(7) Å) is distinctly longer than in the case of sulfoxides (typical bond length: 1.50 Å),13 indicating that 6 exists as the sulfenyl form (R–S–O–H) rather than the sulfoxide form (R–S(O)–H). Although several crystal structures of Cys–SOH in proteins have been characterized,4 the resolution has been limited to ca. 1.7 Å. The crystal structure of 6 allows detailed discussion of the structural parameters of Cys–SOH. This structural information is expected to serve as the model for the geometric adjustment of a Cys–SOH moiety in protein crystallographic analysis.
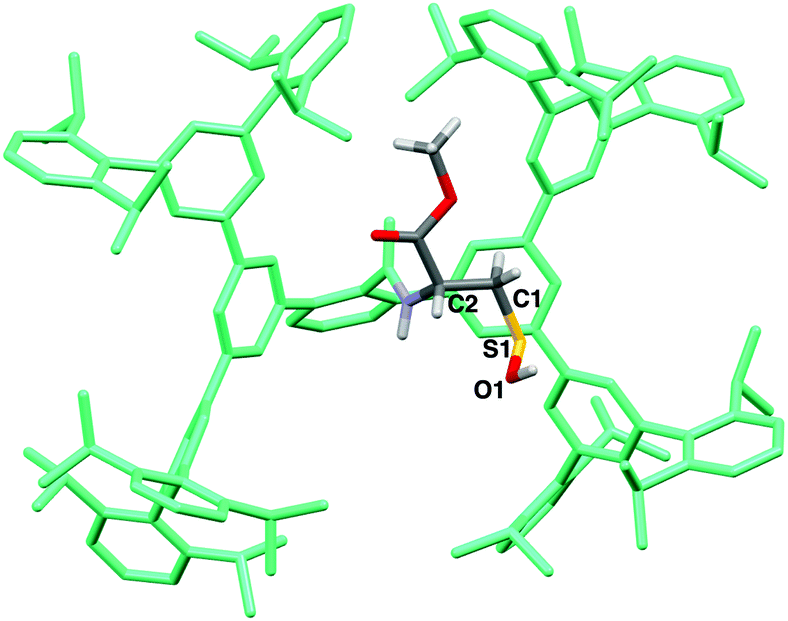 | ||
| Fig. 2 Crystal structure of Cys–SOH 6. Selected bond lengths (Å), bond angle (deg), and torsion angle (deg): S(1)–O(1) 1.616(7); C(1)–S(1) 1.799(8); C(1)–S(1)–O(1) 99.5(4); O(1)–S(1)–C(1)–C(2) –59.9(7). | ||
Cys–SOH 6 exhibited remarkable thermal stability, both in the solid state and in solution. In the solid state, it decomposed at a high temperature of 214 °C. In solution, no detectable change was observed in the 1H NMR spectrum of 6 in C6D6 at 50 °C over 17 h. It remained intact in the presence of triethylamine in THF at room temperature for 7 days.
The reaction of Cys–SOH with a thiol to produce a disulfide is one of the most fundamental processes in redox regulation and protein folding.14 However, the chemical information for this process has been accumulated in a rather indirect fashion, that is, without direct observation of the reacting Cys–SOH. In the studies on non-cysteinyl R–SOH, there are only a few reports on the reaction of stable R–SOH with a thiol.7a,c,e In these examples, the corresponding disulfides were obtained in good yields, but the reactions were found to be extremely slow. The reactivity of Cys–SOH 6 toward a thiol was then investigated. When 6 was treated with N-acetylcysteine methyl ester (7) in THF/H2O (10:1 v/v) at room temperature, the corresponding mixed disulfide 8 was formed as the sole product, but the conversion of 6 to 8 was only 54% after 15 days (Scheme 3a). The addition of benzoic acid as an acid catalyst had virtually no accelerating effect (Scheme 3b). In sharp contrast, the reaction was strongly enhanced in the presence of triethylamine and completed within 10 min, to afford 8 quantitatively (Scheme 3c). Since 8 was immediately formed in the latter case, it is unlikely that the sluggish reaction in the former two cases is due to the steric hindrance caused by the Bpsc cradle. In the latter case, triethylamine is considered to play crucial roles both in increasing the nucleophilicity of the thiol and in increasing the leaving ability of the OH group. The triethylammonium ion generated in the deprotonation of a thiol to form a thiolate will behave as an acid catalyst through protonation of the OH group. These results indicate that, without an appropriate additive, the reaction of Cys–SOH with a thiol is intrinsically slow, suggesting the importance of a basic residue in the vicinity of the –SOH functionality for facile disulfide formation in biological systems.
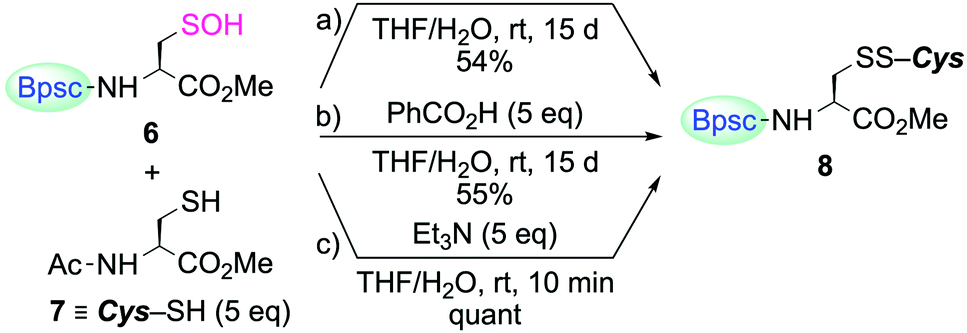 | ||
| Scheme 3 Reaction of Cys–SOH 6 with thiol 7. The yields of 8 were estimated by 1H NMR spectroscopy. | ||
The cradled Cys–SOH 6 was reduced by DTT in the presence of triethylamine to afford Cys–SH 5 in 85% yield (Scheme 4), demonstrating the reversible redox processes between Cys–SH and Cys–SOH. The mixed disulfide 8 was also reduced to 5 under similar conditions.
 | ||
| Scheme 4 Reduction of Cys–SOH 6 to Cys–SH 5. | ||
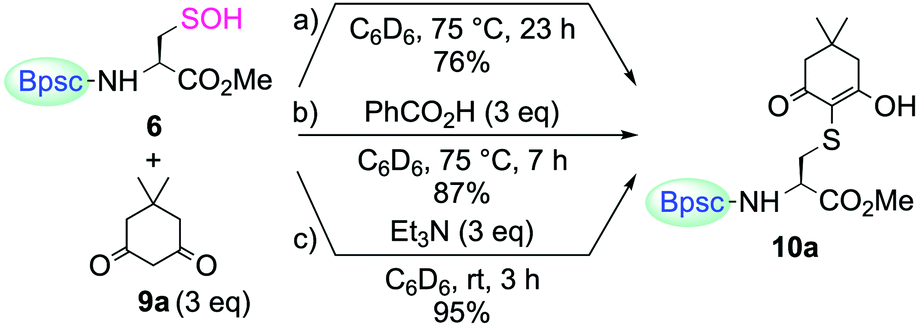
For trapping Cys–SOH generated in proteins, various types of chemical probes have been developed, based on the electrophilic15 and nucleophilic16 reactivities of Cys–SOH. Among them, 5,5-dimethyl-1,3-cyclohexanedione (9a, dimedone) is the most commonly and widely utilized chemical probe for Cys–SOH. A variety of derivatives based on the cyclic 1,3-dicarbonyl scaffold have also been developed. Recently, Carroll et al. reported the preparation of a library of dimedone-type chemical probes and the screening of their properties by evaluating the reactivity toward Cys–SOH, generated in situ by hydrolysis of a dipeptide-based cyclic sulfenamide.5a,15c–e Whereas studies on the chemical probes for Cys–SOH have been carried out exclusively with in situ-generated Cys–SOH, or transiently generated species, isolable Cys–SOH models that can be utilized under reaction conditions different from those used for its generation would serve the development of such studies by removing various constraints in conducting the experiments. The reaction of 6 with dimedone (9a) under various conditions was then investigated.
When 6 was treated with 9a in C6D6, without any additives, the enol form of the corresponding S-adduct 10a was obtained as the sole product (Scheme 5a). However, the reaction was extremely slow. After heating at 75 °C for 23 h, the yield of 10a was 76%. The addition of benzoic acid slightly accelerated the reaction, although heating was still necessary (87% yield after heating at 75 °C for 7 h) (Scheme 5b). In stark contrast, the reaction was remarkably enhanced in the presence of triethylamine; it was completed within 3 h at room temperature to afford 10a in 95% yield (Scheme 5c). Just as in the case of the reaction of 6 with a thiol, the amine base is considered to play important roles in increasing the nucleophilicity of 9a and the leaving ability of the hydroxy group of 6. In their study, utilizing a dipeptide-based sulfenic acid generated in situ, Carroll et al. showed that the reaction of 9a with the sulfenic acid is pH-dependent and that the reaction rate is decreased in the low pH region.15c The result of the present study, that the reaction of 9a with 6 required the addition of an amine to proceed smoothly, is consistent with their results, corroborating the findings that dimedone (9a) can trap a sulfenic acid efficiently only under appropriate reaction conditions.
 | ||
| Scheme 5 Reaction of Cys–SOH 6 with dimedone 9a. | ||
The reactivity of cyclohexane-1,3-dione (9b) and 1,3-dimethylbarbituric acid (9c) toward 6 was also determined (Scheme 6). When 6 was treated with 9b or 9c in the presence of triethylamine in C6D6 at room temperature, the enol forms of the corresponding S-adduct 10b or 10c were obtained in good yields, respectively.
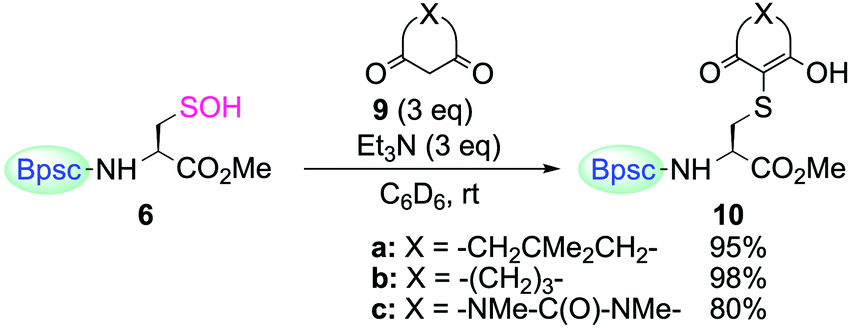 | ||
| Scheme 6 Reaction of Cys–SOH 6 with 1,3-dicarbonyl compounds 9a–c. Reaction time for (a) 3 h, (b) 6.5 h, and (c) 2.5 h. | ||
The kinetic study of the reaction of 6 with 9a (3 equiv.) in the presence of triethylamine (3 equiv.) at room temperature provided a second-order rate constant value of k = 6.8 M−1 s−1. For the reaction of 6 with 9b and 9c, second-order rate constants of k = 6.3 and 8.3 M−1 s−1, respectively, were determined. It is notable that the rate constant for the reaction of 6 with 9a (k = 6.8 M−1 s−1) is of the same order as that of the in situ-generated dipeptide-based sulfenic acid reported by Carroll et al. (k = 11.8 M−1 s−1, in 2:1 PBS buffer:ACN, pH = 7.4, r.t.),15c although there are differences in the solvents and reaction conditions. These results suggest that the reactivity of 6 is not unduly hindered by the Bpsc cradle. In this cradled cysteine model, the –SOH functionality is peripherally protected, yet sufficiently accessible, to appropriate reagents.
In conclusion, the first synthesis and isolation of a small-molecule Cys–SOH has been achieved by taking advantage of a cradled cysteine model and its structure was established by X-ray crystallographic analysis. The cradled Cys–SOH, having both high stability and sufficient reactivity, is expected to serve as a biorepresentative small-molecule sulfenic acid model in research efforts to realize a better understanding the chemical behavior of Cys–SOH in biological systems. Further investigations on its reactivity towards various biologically relevant reagents as well as on the development of its water-soluble derivative are currently in progress.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) W. S. Allison, Acc. Chem. Res., 1976, 9, 293–299 CrossRef CAS; (b) L. B. Poole, P. A. Karplus and A. Claiborne, Annu. Rev. Pharmacol. Toxicol., 2004, 44, 325–347 CrossRef CAS; (c) L. B. Poole and K. J. Nelson, Curr. Opin. Chem. Biol., 2008, 12, 18–24 CrossRef CAS; (d) C. E. Paulsen and K. S. Carroll, ACS Chem. Biol., 2010, 5, 47–62 CrossRef CAS; (e) C. E. Paulsen and K. S. Carroll, Chem. Rev., 2013, 113, 4633–4679 CrossRef CAS; (f) V. Gupta and K. S. Carroll, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 847–875 CrossRef CAS; (g) D. W. Bak and E. Weerapana, Mol. BioSyst., 2015, 11, 678–697 RSC; (h) N. O. Devarie-Baez, E. I. S. Lopez and C. M. Furdui, Free Radical Res., 2016, 50, 172–194 CrossRef CAS.
- (a) E. Block, Garlic and Other Alliums: The Lore and the Science, RSC, Cambridge, 2010, pp. 100–223 Search PubMed; (b) E. Block, J. Z. Gillies, C. W. Gillies, A. A. Bazzi, D. Putman, L. K. Revelle, D. Y. Wang and X. Zhang, J. Am. Chem. Soc., 1996, 118, 7492–7501 CrossRef CAS; (c) L. A. G. M. Van den Broek, L. P. C. Delbressine and H. C. J. Ottenheijm, in The Chemistry of Sulfenic Acids and Their Derivatives, ed. S. Patai, Wiley, New York, 1990, pp. 701–721 Search PubMed.
- (a) D. R. Hogg, in The Chemistry of Sulfenic Acids and Their Derivatives, ed. S. Patai, Wiley, New York, 1990, pp. 361–402 Search PubMed; (b) A. L. Schwan and S. C. Soderman, Phosphorus Sulfur Silicon Relat. Elem., 2013, 188, 275–286 CrossRef CAS; (c) E. Block, J. Sulfur Chem., 2013, 34, 158–207 CrossRef CAS.
- For examples, see: (a) J. I. Yeh, A. Claiborne and W. G. J. Hol, Biochemistry, 1996, 35, 9951–9957 CrossRef CAS; (b) H. J. Choi, S. W. Kang, C. H. Yang, S. G. Rhee and S. E. Ryu, Nat. Struct. Biol., 1998, 5, 400–406 CrossRef CAS; (c) R. L. M. van Montfort, M. Congreve, D. Tisi, R. Carr and H. Jhoti, Nature, 2003, 423, 773–777 CrossRef CAS; (d) T. Nakamura, T. Yamamoto, M. Abe, H. Matsumura, Y. Hagihara, T. Goto, T. Yamaguchi and T. Inoue, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6238–6242 CrossRef CAS; (e) C. B. Poor, P. R. Chen, E. Duguid, P. A. Rice and C. He, J. Biol. Chem., 2009, 284, 23517–23524 CrossRef CAS.
- For recent reviews, see: (a) Y. Shi and K. S. Carroll, Acc. Chem. Res., 2020, 53, 20–31 CrossRef CAS; (b) J. M. M. Pople and J. M. Chalker, Curr. Opin. Chem. Biol., 2021, 60, 55–65 CrossRef CAS.
- For kinetically stabilized sulfenic acids, see: (a) N. Nakamura, J. Am. Chem. Soc., 1983, 105, 7172–7173 CrossRef CAS; (b) T. Yoshimura, E. Tsukurimichi, S. Yamazaki, S. Soga, C. Shimasaki and K. Hasegawa, J. Chem. Soc., Chem. Commun., 1992, 1337–1338 RSC; (c) A. Ishii, K. Komiya and J. Nakayama, J. Am. Chem. Soc., 1996, 118, 12836–12837 CrossRef CAS; (d) M. Yukimoto, R. Nishino, F. Suzuki, M. Ishihara, K. Sugamata and M. Minoura, Chem. Lett., 2018, 47, 425–428 CrossRef CAS.
- For our previous reports on kinetically stabilized sulfenic acids, see: (a) K. Goto, N. Tokitoh and R. Okazaki, Angew. Chem., Int. Ed. Engl., 1995, 34, 1124–1126 CrossRef CAS; (b) T. Saiki, K. Goto, N. Tokitoh and R. Okazaki, J. Org. Chem., 1996, 61, 2924–2925 CrossRef CAS; (c) K. Goto, M. Holler and R. Okazaki, J. Am. Chem. Soc., 1997, 119, 1460–1461 CrossRef CAS; (d) K. Goto, K. Shimada, S. Furukawa, S. Miyasaka, Y. Takahashi and T. Kawashima, Chem. Lett., 2006, 35, 862–863 CrossRef CAS; (e) M. Ishihara, N. Abe, S. Sase and K. Goto, Chem. Lett., 2015, 44, 615–617 CrossRef CAS.
- For thermodynamically stabilized sulfenic acids, see: (a) K. Fries, Ber. Dtsch. Chem. Ges., 1912, 45, 2965–2973 CrossRef; (b) B. C. Pal, M. Uziel, D. G. Doherty and W. E. Cohn, J. Am. Chem. Soc., 1969, 91, 3634–3638 CrossRef CAS; (c) A. Heckel and W. Pfleiderer, Tetrahedron Lett., 1983, 24, 5047–5050 CrossRef CAS; (d) R. Tripolt, F. Belaj and E. Nachbaur, Z. Naturforsch., B: J. Chem. Sci., 1993, 48, 1212–1222 CAS; (e) T. Machiguchi, T. Hasegawa, H. Otani, S. Yamabe and H. Mizuno, J. Am. Chem. Soc., 1994, 116, 407–408 CrossRef CAS; (f) T. Fekner, J. E. Baldwin, R. M. Adlington and C. J. Schofield, Tetrahedron Lett., 1998, 39, 6983–6986 CrossRef CAS; (g) X. B. Li, Z. F. Xu, L. J. Liu and J. T. Liu, Eur. J. Org. Chem., 2014, 1182–1188 CrossRef CAS.
- For recent examples, see: (a) K. Goto, D. Sonoda, K. Shimada, S. Sase and T. Kawashima, Angew. Chem., Int. Ed., 2010, 49, 545–547 CrossRef CAS; (b) S. Sase, R. Kakimoto and K. Goto, Angew. Chem., Int. Ed., 2015, 54, 901–904 CrossRef CAS; (c) T. Sano, K. Shimada, Y. Aoki, T. Kawashima, S. Sase and K. Goto, Molecules, 2017, 22, 19 CrossRef; (d) S. Sase, R. Kimura, R. Masuda and K. Goto, New J. Chem., 2019, 43, 6830–6833 RSC.
- For a review, see: K. Goto, in Synergy in Supramolecular Chemistry, ed. T. Nabeshima, CRC Press, Boca Raton, 2015, pp. 191–218 Search PubMed.
- (a) H. Z. Chae, T. B. Uhm and S. G. Rhee, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 7022–7026 CrossRef CAS; (b) H. R. Ellis and L. B. Poole, Biochemistry, 1997, 36, 13349–13356 CrossRef CAS; (c) J. M. Denu and K. G. Tanner, Biochemistry, 1998, 37, 5633–5642 CrossRef CAS; (d) C. C. Winterbourn and D. Metodiewa, Free Radical Biol. Med., 1999, 27, 322–328 CrossRef CAS; (e) L. M. S. Baker and L. B. Poole, J. Biol. Chem., 2003, 278, 9203–9211 CrossRef CAS; (f) J. J. Tanner, Z. D. Parsons, A. H. Cummings, H.-Y. Zhou and K. S. Gates, Antioxid. Redox Signaling, 2011, 15, 77–97 CrossRef CAS.
- J.-P. R. Chauvin and D. A. Pratt, Angew. Chem., Int. Ed., 2017, 56, 6255–6259 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- (a) D. S. Rehder and C. R. Borges, Biochemistry, 2010, 49, 7748–7755 CrossRef CAS; (b) B. G. Hill and A. Bhatnagar, J. Mol. Cell. Cardiol., 2012, 52, 559–567 CrossRef CAS; (c) A. Zeida, M. Trujillo, G. Ferrer-Sueta, A. Denicola, D. A. Estrin and R. Radi, Chem. Rev., 2019, 119, 10829–10855 CrossRef CAS; (d) M. A. Hagras, M. A. Bellucci, G. Gobbo, R. A. Marek and B. L. Trout, J. Phys. Chem. B, 2020, 124, 9840–9851 CrossRef CAS.
- For examples, see: (a) L. V. Benitez and W. S. Allison, J. Biol. Chem., 1974, 249, 6234–6243 CrossRef CAS; (b) C. Klomsiri, K. J. Nelson, E. Bechtold, L. Soito, L. C. Johnson, W. T. Lowther, S.-E. Ryu, S. B. King, C. M. Furdui and L. B. Poole, Methods Enzymol., 2010, 473, 77–94 CAS; (c) V. Gupta and K. S. Carroll, Chem. Sci., 2016, 7, 400–415 RSC; (d) V. Gupta and K. S. Carroll, Chem. Commun., 2016, 52, 3414–3417 RSC; (e) V. Gupta, H. Paritala and K. S. Carroll, Bioconjugate Chem., 2016, 27, 1411–1418 CrossRef CAS.
- For examples, see: (a) T. H. Poole, J. A. Reisz, W. Zhao, L. B. Poole, C. M. Furdui and S. B. King, J. Am. Chem. Soc., 2014, 136, 6167–6170 CrossRef CAS; (b) D. J. McGarry, M. M. Shchepinova, S. Lilla, R. C. Hartley and M. F. Olson, ACS Chem. Biol., 2016, 11, 3300–3304 CrossRef CAS; (c) L. J. Alcock, B. L. Oliveira, M. J. Deery, T. L. Pukala, M. V. Perkins, G. J. L. Bernardes and J. M. Chalker, ACS Chem. Biol., 2019, 14, 594–598 CrossRef CAS; (d) S. L. Scinto, O. Ekanayake, U. Seneviratne, J. E. Pigga, S. J. Boyd, M. T. Taylor, J. Liu, C. W. am Ende, S. Rozovsky and J. M. Fox, J. Am. Chem. Soc., 2019, 141, 10932–10937 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2044613. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc08422k |
| This journal is © The Royal Society of Chemistry 2021 |