
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeting SARS-CoV-2 spike protein by stapled hACE2 peptides†
Marijn N.
Maas
 ,
Jordi C. J.
Hintzen
,
Philipp M. G.
Löffler
and
Jasmin
Mecinović
*
,
Jordi C. J.
Hintzen
,
Philipp M. G.
Löffler
and
Jasmin
Mecinović
*
University of Southern Denmark, Department of Physics, Chemistry and Pharmacy, Campusvej 55, 5230 Odense, Denmark. E-mail: mecinovic@sdu.dk; Tel: +45-6550-3603
First published on 2nd March 2021
Abstract
SARS-CoV-2 Spike protein RBD interacts with the hACE2 receptor to initiate cell entry and infection. We set out to develop lactam-based i,i + 4 stapled hACE2 peptides targeting SARS-CoV-2. In vitro screening demonstrates the inhibition of the Spike protein RBD-hACE2 complex formation by the hACE221-55A36K-F40E stapled peptide (IC50: 3.6 μM, Kd: 2.1 μM), suggesting that hACE2 peptidomimetics could form the basis for the development of anti-COVID-19 therapeutics.
The global COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, or 2019-nCoV) has rapidly introduced worldwide health concerns and economic challenges of unprecedented scale.1,2 As of February 2021, the COVID-19 outbreak has been responsible for over 100 million infections.3 Many nations are facing a second wave of infections, which results in further strain on public healthcare. The lack of approved antiviral agents specifically targeting SARS-CoV-2 has driven a collective shift towards global R&D efforts aimed at developing therapeutics that act against COVID-19.4
Whole-genome sequencing and subsequent phylogenetic analysis of SARS-CoV-2 led to the classification of the virion as a member of the beta coronavirus genus.5,6 Members of the coronavirus genus encode four structural proteins on their positive sense, single stranded RNA ((+)ssRNA);7 S (spike), E (envelope), M (membrane), and N (nucleocapsid).8 The common denominator between the coronavirus strains resulting in human pathogenicity is the viral S glycoprotein trimer. These trans-membrane glycoproteins selectively bind the protease domain (PD) of host cell surface peptidases with a receptor-binding domain (RBD) to mediate virion-membrane fusion to initiate host infection.9–11 SARS-CoV-2 specifically targets the transmembrane human angiotensin-converting enzyme 2 (hACE2) for cell entry (Fig. 1a).12 Structural analyses of the SARS-CoV-2-hACE2 complex demonstrate that the RBD facilitates protein–protein interactions (PPI) to the α1-helix of hACE2 (Fig. 1a). Key binding contributions are provided by the α1-helix (Fig. 1b), while minor binding contributions are maintained by the α2-helix, and β3- and β4-sheet linker of hACE2 (Fig. 1a).13–16 Interference of this PPI interface could potentially prevent SARS-CoV-2 from entering the human cells via association with hACE2, inhibiting subsequent viral replication.17,18
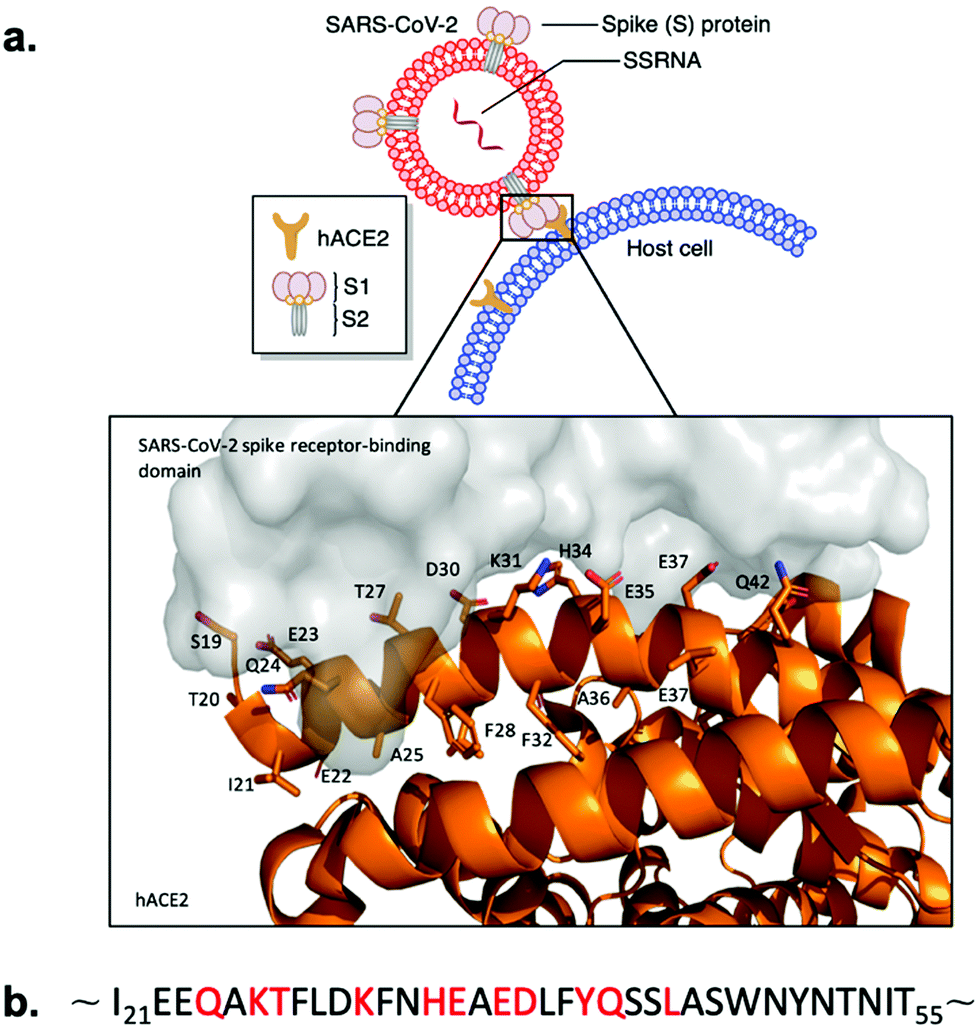 | ||
| Fig. 1 SARS-CoV-2 Spike protein RBD binding to hACE2. (a) Schematic presentation and crystal structure of SARS-CoV-2 Spike protein RBD (grey) binding to the extracellular domain of hACE2 (orange) (PDB ID: 6M0J). Important residues of the hACE2 N-terminal α1-helix are labeled. (b) Sequence of the hACE2 N-terminal α1-helix. Key binding residues are marked with red. | ||
Various small-molecule compounds were predicted to target the binding interface between SARS-CoV-2 S protein RBD and hACE2 through computational studies.19–21 Experimental work by Carino et al. demonstrated that small-molecules could lead to PPI inhibition between Spike RBD and the carboxypeptidase domain of hACE2 with natural and semi synthetic steroidal agents.19 However, small-molecules targeting a large surface area of the RBD cannot engage with the entire extended linear ridge-like binding region.8
Peptidomimetics are better suited for the inhibition of PPI compared to small molecules due to efficient engagement with large protein surfaces through entropically favored interactions with protein surfaces, preventing competitive replacement.22,23 hACE2 α1-helix-based peptidomimetics targeting SARS-CoV-2 S protein were demonstrated to inhibit RBD-hACE2 complex formation and subsequent host cell infection in both computational and experimental studies.24–26 However, hACE2 α1-helix-based peptides are expected to lose their bio-active conformation in solution, decreasing the RBD binding potential of the peptidomimetic.17 Furthermore, short and linear peptide-based drugs are generally cell impermeable, not orally available, and subject to rapid proteolysis.27,28 These limitations can be overcome with i,i + 4 peptide stapling approaches that stabilise the peptide's three-dimensional and bio-active structure, increase cellular uptake, decrease enzymatic degradation and improve pharmacokinetic properties through chemical modification of the peptide sequence.27,29 However, somewhat conflicting recent reports showed that hydrocarbon-based stapling of hACE2 might only partially lead to the disruption of the RBD-hACE2 complex.30,31 In addition, most SARS-CoV-2 replication takes place in the lower and upper airways, which potentially allows for targeting of the RBD-hACE2 interaction by inhalation of the aerosolised peptide therapeutics. This makes the potential lack of oral availability of peptides a non-issue.32
We hypothesised that the development of a stapled hACE2 N-terminal α1-helix might allow for selective and potent inhibition of RBD-hACE2 recognition and subsequent infection. In this report we discuss the design and synthesis of lactam-based stapled 35-mer peptides using the hACE2 N-terminal α1-helix inhibitor 1 sequence to overcome structural instability and increase peptide binding affinity to SARS-CoV-2 (Fig. 2).
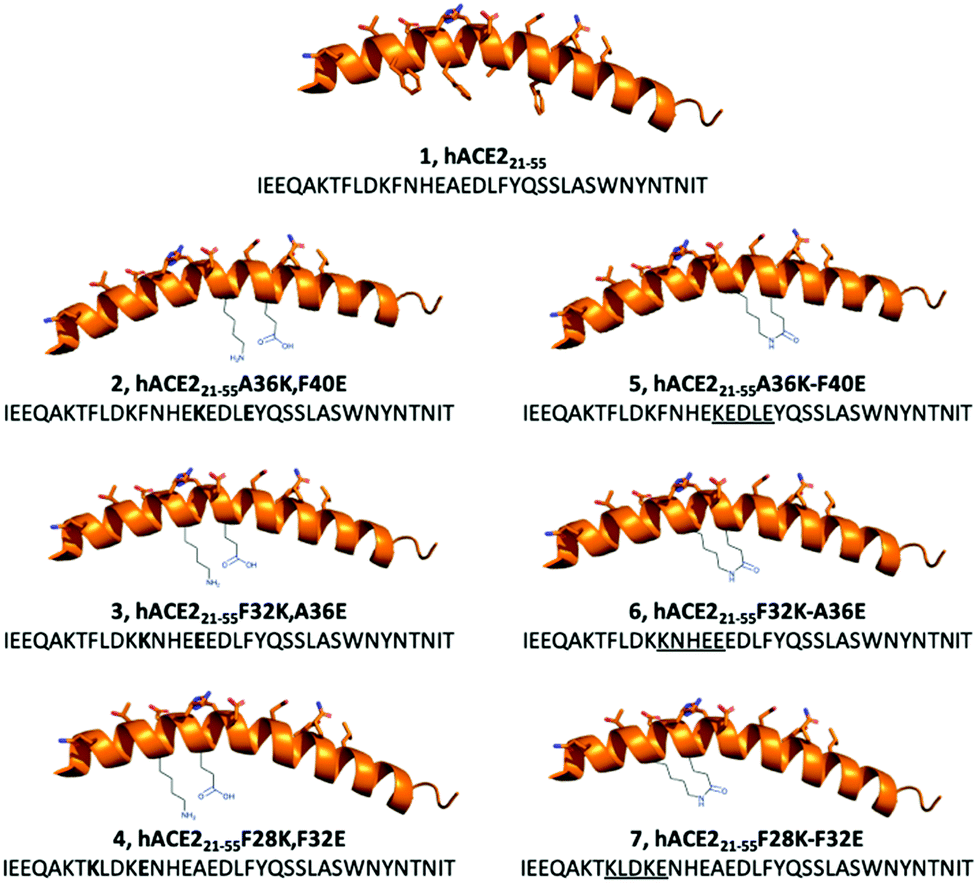 | ||
| Fig. 2 Design of lactam-based i,i + 4 stapled hACE2-based peptides for the inhibition of SARS-CoV-2 S protein-hACE2 interactions. The wild-type sequence (1) and linear modified peptides (2–4) were used as controls to their stapled counterparts (5–7). | ||
Cryo-EM and X-ray structures, as well as computational analysis of the interaction between hACE2 and RBD revealed that residues 21–55 of the N-terminal α1-helix of hACE2 act as the major contributors of binding interactions with S protein RBD (Fig. 1). Král et al. computationally determined that several polar residues of the N-terminal α1-helix are essential mediators of RBD binding (Fig. 1b).20 We selected peptide i,i + 4 stapling sites (F28-F32, F32-A36, A36-F40) based on their positioning in the hACE2 N-terminal α1-helix structure to not interfere with the binding interface (Fig. 1b).17,20 Lactam-linked stapling was shown to effectively induce helicity, uses natural building blocks, and facilitates straightforward synthesis.33
35-mer native peptide 1 and linear controls 2–4 were synthesised by solid-phase peptide synthesis (SPPS) (Fig. 2). Stapled peptides 5–7 were synthesised using SPPS, followed by stapling of the side-chains with lactam-based i,i + 4 methods, and cleavage of the stapled hACE2 peptides from the resin (Fig. 2). Mass spectrometry and analytical HPLC confirmed the high purity (>90%) of all RP-HPLC purified synthetic hACE2 peptides (Table S1 and Fig. S1–S7, ESI†).
α-Helical propensity of the linear hACE2 peptides could be computationally predicted with the use of the PSIPRED secondary structure prediction server (Fig. S8, ESI†).34 α-Helical content of the synthetic hACE2 peptides was confirmed by CD spectroscopy to determine the effect of i,i + 4 lactam stapling on the peptide secondary structure with the expectation that stapling increases peptide helicity, using poly-L-lysine as a reference for helicity (Fig. 3 and Fig. S9, ESI†).35,36 Spectra were recorded in 10 mM PBS at pH 7.4 in the absence and the presence of 2,2,2-trifluoroethanol (TFE) at 25 °C (Fig. 3). TFE is known to stimulate the formation of the α-helical secondary structures in peptides,37 allowing for a comparison of the α-helical content of hACE2 peptides at maximal helicity (Fractional Helicity, fH), which we observed at 30% TFE (Fig. S9, ESI†). CD spectra were deconvoluted with the DICHROWEB server to determine helicities.38 CD spectroscopy demonstrated low helicity for both linear and stapled hACE2 peptides in absence of TFE, with predictors of helicity averaging to 6-13% helical content (Table S2 and Fig. S10, ESI†). A lack of the predominant α-helical secondary structure of the natural hACE2 peptide sequence in the absence of TFE is in agreement with findings by Karoyan et al. (Table S2, ESI†).24 In the presence of TFE, however, α-helical structures can be observed for various synthetic hACE2 peptides with predictors averaging from 11 to 52% helical content, with a clear shift in negative ellipticity from 200 to 208 and 222 nm at varying intensities for all peptides (Fig. 3, Fig. S9, S10, Table S2, ESI†). Stapled peptide 5 displayed the highest helicity across the panel of hACE2 peptides (average: 52%), with increased helical content compared to the wild-type sequence 1 (average: 38%). Stapling closer to the N-terminus seems to result in a loss of helicity compared to the native sequence. This observation suggests that i,i + 4 lactam stapling at the A36K-F40E site can positively affect the helical content, possibly due to the sequence environment effects surrounding the peptide staple.
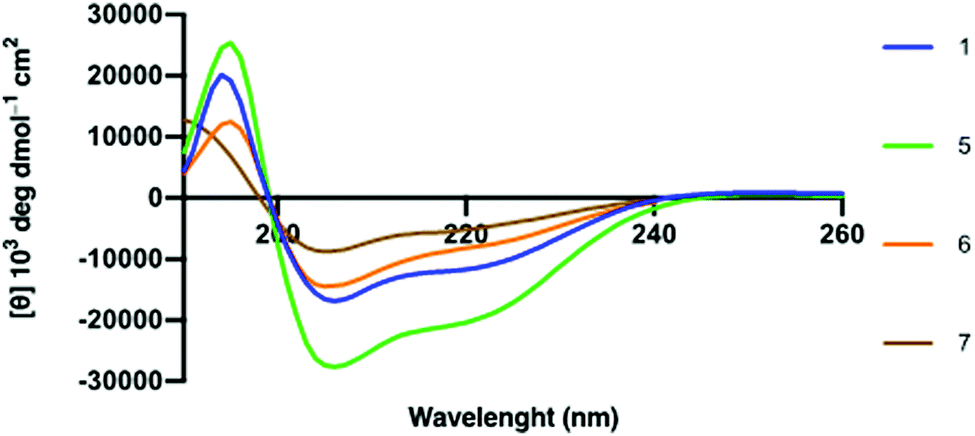 | ||
| Fig. 3 Circular Dichroism spectra of hACE2 peptides in molar ellipticity per residue. 30 μM peptide in 10 mM PBS at pH 7.4 with 30% TFE at 298 K. | ||
Synthetic hACE2 peptides were evaluated as inhibitors of SARS-CoV-2 Spike protein-hACE2 complex formation using the ELISA based Screening Assay Kit. A concentration–response curve of SARS-CoV-2 Spike Protein (RBD) Recombinant Human Monoclonal Antibody in PBS was used as a positive control for inhibition (0.6 nM–200 μM) and to test the viability of the ELISA kit (Fig. S11, ESI†). A concentration-dependent decrease of luminescence was observed along with a median inhibitory concentration (IC50) for the monoclonal antibody (9.5 nM), similar to the reported IC50 value for the antibody (6.6 nM) targeting SARS-CoV-2 Spike protein by the manufacturer (Fig. S11, ESI†). We evaluated the linear and stapled hACE2 peptides at 10 μM and 100 μM to identify potent inhibitors of the RBD-hACE2 complex formation (Fig. 4a). Among all peptides, only 5 reduced binding by >80% at 10 μM. Concentration-dependent assays (0.3–300 μM) of the stapled hACE2 peptides demonstrated significant inhibition of binding by 5 (IC50: 3.6 μM) compared to the linear wild-type control 1 that displayed only minor inhibition at >100 μM concentrations (Fig. 4b, n = 3 replicates). Peptides 6 (IC50: 28.4 μM) and 7 (IC50: 46.8 μM) displayed enhanced inhibitory activity compared to the native sequence, although 12–18 times less potent when compared to 5 (Fig. S11, ESI†). The linear controls 2–4 showed reduced binding to 50% at 100 μM, suggesting that the linear peptides are poor inhibitors of SARS-CoV-2-hACE2 interactions and that decreased hACE2 helicity results in less potent inhibitory effects (Fig. 4a).
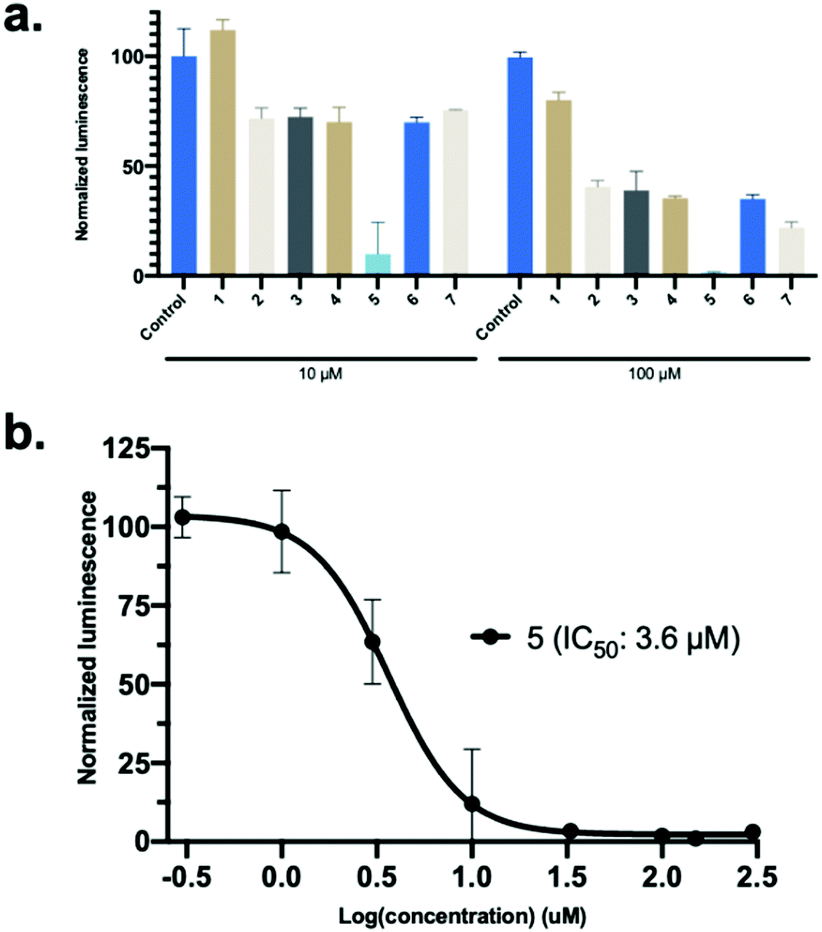 | ||
| Fig. 4 SARS-CoV-2 Spike protein-hACE2 inhibitor screening assay. (a) The linear and stapled hACE2 peptides was screened for inhibition at 10 μM and 100 μM. Data is mean ± SD, n = 2. (b) Concentration-dependent (0.3–300 μM) IC50 determination of 5. Data is mean ± SD, n = 3. | ||
Surface Plasmon Resonance (SPR) was used to quantify the binding affinity (Kd) and kinetic association and dissociation parameters (kon and koff) between SARS-CoV-2 Spike protein and the stapled hACE221-55 peptides (5, 6 and 7) using the polycarboxylate HC surface SPR sensor chip with pre-immobilized SARS-CoV-2 Spike protein with a Biacore X100. Full-length hACE2 protein and 1 were used as positive and negative controls, respectively (Fig. S12, ESI†). The binding interaction between 5 and the immobilized SARS-CoV-2 Spike protein was characterized by micromolar binding affinity (Kd: 2.1 ± 0.2 μM), a fast association rate (kon: 1.7 ± 0.1 × 104 M−1 s−1) and a moderate rate of dissociation (koff: 0.034 ± 0.002 s−1) (Fig. 5 and Table S3, ESI†). The binding affinity of the native sequence 1 and stapled peptides 6 and 7 could not be determined, as the interaction with the Spike protein versus the reference surface was too weak for the assessment (Fig. S13, ESI†). These results, in combination with the inhibition assay, indicate direct and potent binding of hACE221-55A36K-F40E (5) peptide to the RBD domain of Spike protein.
 | ||
| Fig. 5 Determination of binding of stapled hACE2 peptide 5 (0–20 μM) to immobilized SARS-CoV-2 Spike protein with SPR. Conditions: 10 mM sodium phosphate, pH 7.4, 25 °C, n = 2. | ||
Finally, resistance to proteolysis was determined for the 35-mer peptides (1, 2 and 5) by treating the peptides with human serum (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 v/v, serum:PBS) at 37 °C and recording peptide degradation over time using analytical HPLC (Fig. 6). Interestingly, the data demonstrated that both 1 and 5 remain highly stable over time. Peptide 2 was observed to be comparatively less stable than the natural sequence, which is likely due to the introduction of more sensitive proteolytic cleavage sites by substitution of A36K and F40E.
:4 v/v, serum:PBS) at 37 °C and recording peptide degradation over time using analytical HPLC (Fig. 6). Interestingly, the data demonstrated that both 1 and 5 remain highly stable over time. Peptide 2 was observed to be comparatively less stable than the natural sequence, which is likely due to the introduction of more sensitive proteolytic cleavage sites by substitution of A36K and F40E.
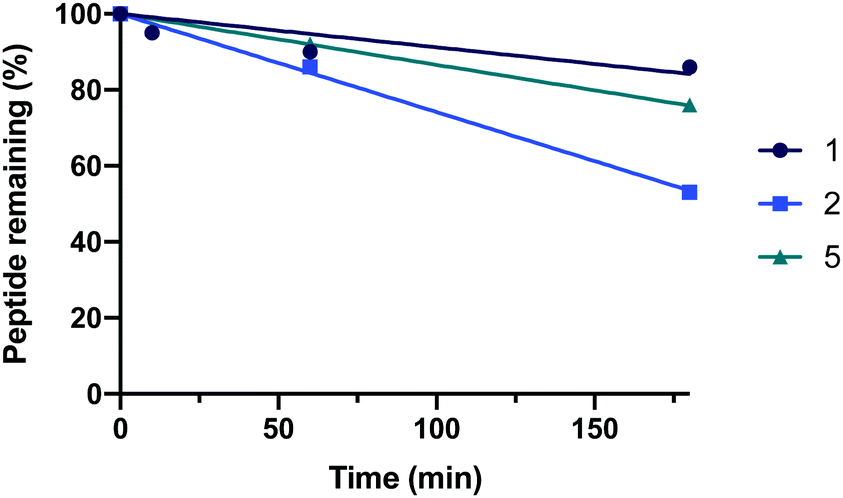 | ||
| Fig. 6 The serum stability of hACE2 peptides 1, 2 and 5. | ||
In conclusion, we have developed lactam-based i,i + 4 stapled hACE2 α1-helix-based peptidomimetics that target SARS-CoV-2 S protein and inhibit the RBD-hACE2 complex formation. We envision that hACE2 α1-helix-based peptidomimetics could potentially prevent SARS-CoV-2 from entering the human cells through hACE2 and thus inhibit subsequent viral replication. Our work highlights that the stapled peptide hACE221-55A36K-F40E (5), which exhibits an increased level of helicity, efficiently inhibits SARS-CoV-2 Spike protein-hACE2 binding (IC50: 3.6 μM, Kd: 2.1 ± 0.2 μM). Related stapled peptides hACE221-55F32K-A36E (6) and hACE221-55F28K-F32E (7) also resulted in stronger inhibition compared to the natural sequence 1. Taken together, we believe that continued optimisation of the chosen staples and additional sequence modifications might contribute to the design of future anti-coronavirus therapeutics, which can aid with infection prevention.
This work was supported by the ERC Starting Grant to J. M. (ChemEpigen-715691). P. M. G. L. thanks the Villum Fonden (BioNEC-VKR18333). We thank the Danish Molecular Biomedical Imaging Centre (University of Southern Denmark) for the use of bioimaging facilities.
Conflicts of interest
There are no conflicts to declare.References
- F. Wu, S. Zhao, B. Yu, Y. M. Chen, W. Wang, Z. G. Song, Y. Hu, Z. W. Tao, J. H. Tian, Y. Y. Pei, M. L. Yuan, Y. L. Zhang, F. H. Dai, Y. Liu, Q. M. Wang, J. J. Zheng, L. Xu, E. C. Holmes and Y. Z. Zhang, Nature, 2020, 579, 265–269 CrossRef CAS.
- F. Jiang, L. Deng, L. Zhang, Y. Cai, C. W. Cheung and Z. Xia, J. Gen. Intern. Med., 2020, 35, 1545–1549 CrossRef.
- Worldometer, Coronavirus Cases, https://www.worldometers.info/coronavirus/coronavirus-cases/#daily-cases, accessed 8 feb 2021.
- T. Thanh, L. Z. Andreadakis, A. Kumar, R. Gómez Román, S. Tollefsen, M. Saville and S. Mayhew, Nat. Rev. Drug Discovery, 2020, 19, 305–306 CrossRef.
- P. C. Y. Woo, S. K. P. Lau, C. S. F. Lam, K. K. Y. Lai, Y. Huang, P. Lee, G. S. M. Luk, K. C. Dyrting, K.-H. Chan and K.-Y. Yuen, J. Virol., 2009, 83, 908–917 CrossRef CAS.
- R. Lu, X. Zhao, J. Li, P. Niu, B. Yang, H. Wu, W. Wang, H. Song, B. Huang, N. Zhu, Y. Bi, X. Ma, F. Zhan, L. Wang, T. Hu, H. Zhou, Z. Hu, W. Zhou, L. Zhao, J. Chen, Y. Meng, J. Wang, Y. Lin, J. Yuan, Z. Xie, J. Ma, W. J. Liu, D. Wang, W. Xu, E. C. Holmes, G. F. Gao, G. Wu, W. Chen, W. Shi and W. Tan, Lancet, 2020, 395, 565–574 CrossRef CAS.
- G. Lu and D. Liu, Protein Cell, 2012, 803–805 CrossRef.
- C. Wu, Y. Liu, Y. Yang, P. Zhang, W. Zhong, Y. Wang, Q. Wang, Y. Xu, M. Li, X. Li, M. Zheng, L. Chen and H. Li, Acta Pharm. Sin. B, 2020, 10, 766–788 CrossRef CAS.
- F. Li, W. Li, M. Farzan and S. C. Harrison, Science, 2005, 309, 1864–1868 CrossRef CAS.
- Y. Wan, J. Shang, R. Graham, R. S. Baric and F. Li, J. Virol., 2020, 94, e0012720 CrossRef.
- S. VanPatten, M. He, A. Altiti, K. F. Cheng, M. H. Ghanem and Y. Al-Abed, Future Med. Chem., 2020, 12(18), 1647–1656 CrossRef CAS.
- H. Zhang, J. M. Penninger, Y. Li, N. Zhong and A. S. Slutsky, Intensive Care Med., 2020, 46, 586–590 CrossRef CAS.
- Q. Wang, Y. Zhang, L. Wu, S. Niu, C. Song, Z. Zhang, G. Lu, C. Qiao, Y. Hu, K. Y. Yuen, Q. Wang, H. Zhou, J. Yan and J. Qi, Cell, 2020, 181(4), 894–904 CrossRef CAS.
- R. Yan, Y. Zhang, Y. Li, L. Xia, Y. Guo and Q. Zhou, Science, 2020, 367, 1444–1448 CrossRef CAS.
- J. Shang, G. Ye, K. Shi, Y. Wan, C. Luo, H. Aihara, Q. Geng, A. Auerbach and F. Li, Nature, 2020, 581, 221–224 CrossRef CAS.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C. L. Hsieh, O. Abiona, B. S. Graham and J. S. McLellan, Science, 2020, 367, 1260–1263 CrossRef CAS.
- C. Lupala, V. Kumar, X. Li, X.-D. Su and H. Liu, bioRxiv, 2020 DOI:10.1101/2020.05.03.075473.
- S. Xiu, A. Dick, H. Ju, S. Mirzaie, F. Abdi, S. Cocklin, P. Zhan and X. Liu, J. Med. Chem., 2020, 63, 12256–12274 CrossRef CAS.
- A. Carino, F. Moraca, B. Fiorillo, S. Marchianò, V. Sepe, M. Biagioli, C. Finamore, S. Bozza, D. Francisci, E. Distrutti, B. Catalanotti, A. Zampella and S. Fiorucci, Front. Chem., 2020, 8, 572885 CrossRef CAS.
- Y. Han, P. Král and P. Kraí, ACS Nano, 2020, 14, 5143–5147 CrossRef CAS.
- Z. Shang, S. Y. Chan, W. J. Liu, P. Li and W. Huang, ACS Infect. Dis., 2020 DOI:10.1021/acsinfecdis.0c00646.
- D. Bojadzic and P. Buchwald, Curr. Top. Med. Chem., 2018, 18, 674–699 CrossRef CAS.
- A. M. Tharappel, S. K. Samrat, Z. Li and H. Li, ACS Infect. Dis., 2020, 6, 2844–2865 CrossRef CAS.
- P. Karoyan, V. Vieillard, E. Odile, A. Denis, A. Guihot, C.-E. Luyt, L. Gómez-Morales, P. Grondin and O. Lequin, bioRxiv, 2020 DOI:10.1101/2020.08.24.264077.
- R. C. Larue, E. Xing, A. D. Kenney, Y. Zhang, J. A. Tuazon, J. Li, J. S. Yount, P. K. Li and A. Sharma, Bioconjugate Chem., 2021, 32, 215–223 CrossRef CAS.
- G. Zhang, S. Pomplun, A. R. Loftis, A. Loas and B. L. Pentelute, bioRxiv, 2020 DOI:10.1101/2020.03.19.999318.
- L. Nevola and E. Giralt, Chem. Commun., 2015, 51, 3302–3315 RSC.
- B. Meibohm, Pharmaceutical Biotechnology: Fundamentals and Applications, Springer International Publishing, 2019, pp. 99–137 Search PubMed.
- T. A. F. F. Cardote and A. Ciulli, ChemMedChem, 2016, 11, 787–794 CrossRef CAS.
- D. C. Morgan, C. Morris, A. Mahindra, C. M. Blair, G. Tejeda, I. Herbert, M. L. Turnbull, G. Lieber, B. J. Willett, N. Logan, B. Smith, A. B. Tobin, D. Bhella, G. Baillie and A. G. Jamieson, Pept. Sci., 2021, e24217 Search PubMed.
- F. Curreli, S. M. B. Victor, S. Ahmed, A. Drelich, X. Tong, C.-T. K. Tseng, C. D. Hillyer and A. K. Debnath, mBio, 2020, 11, e02451 CrossRef CAS.
- D. Schütz, Y. B. Ruiz-Blanco, J. Münch, F. Kirchhoff, E. Sanchez-Garcia and J. A. Müller, Adv. Drug Delivery Rev., 2020, 167, 47–65 CrossRef.
- A. D. de Araujo, H. N. Hoang, W. M. Kok, F. Diness, P. Gupta, T. A. Hill, R. W. Driver, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed., 2014, 53, 6965–6969 CrossRef CAS.
- D. W. A. Buchan and D. T. Jones, Nucleic Acids Res., 2019, 47, W402–407 CrossRef CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS.
- B. Davidson and G. D. Fasman, Biochemistry, 1967, 6, 1616–1629 CrossRef CAS.
- F. D. Sönnichsen, J. E. Van Eyk, R. S. Hodges and B. D. Sykes, Biochemistry, 1992, 31, 8790–8798 CrossRef.
- L. Whitmore and B. A. Wallace, Nucleic Acids Res., 2004, 32, W668–W673 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of hACE2 peptides, CD data, ELISA assays, SPR assays, serum stability assay. See DOI: 10.1039/d0cc08387a |
| This journal is © The Royal Society of Chemistry 2021 |