
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple Hückel model-driven strategy to overcome electronic barriers to retro-Brook silylation relevant to aryne and bisaryne precursor synthesis†
Edward A.
Neal‡
 *,
A. Yannic R.
Werling‡
and
Christopher R.
Jones
*
*,
A. Yannic R.
Werling‡
and
Christopher R.
Jones
*
School of Biological and Chemical Sciences, Queen Mary University of London, Mile End Road, London, UK. E-mail: e.a.neal@qmul.ac.uk; c.jones@qmul.ac.uk
First published on 13th January 2021
Abstract
ortho-Silylaryl triflate precursors (oSATs) have been responsible for many recent advances in aryne chemistry and are most commonly accessed from the corresponding 2-bromophenol. A retro-Brook O- to C-silyl transfer is a key step in this synthesis but not all aromatic species are amenable to the transformation, with no functionalized bisbenzyne oSATs reported. Simple Hückel models are presented which show that the calculated aromaticity at the brominated position is an accurate predictor of successful retro-Brook reaction, validated synthetically by a new success and a predicted failure. From this, the synthesis of a novel difunctionalized bisaryne precursor has been tested, requiring different approaches to install the two C-silyl groups. The first successful use of a disubstituted o-silylaryl sulfonate bisbenzyne precursor in Diels–Alder reactions is then shown.
Arynes are a class of highly reactive electron-deficient intermediate derived from an arene or heteroarene with two vacant positions, usually ortho to each other. The reactions of arynes generally involve the introduction or extension of a π-system and have been extensively reviewed.1 These transformations are particularly useful in the pharmaceuticals industry and in π-extended polyaromatic hydrocarbon (PAH) material production for sensing, energy generation and other applications.1a,c
Classical methods of aryne generation typically required the use of strong bases, sodium metal, pyrophoric tert-butyllithium or potentially explosive ortho-diazocarboxylate precursors, as well as more extreme temperatures. In contrast, ortho-silylaryl triflate precursors (oSATs) operate under milder reaction conditions and possess a more attractive safety profile,2a which has led to their widespread uptake.1,2b–d Originally developed by Kobayashi in 1983,3ao-(trimethylsilyl)phenyl triflate 5 reliably affords benzyne upon treatment with either fluoride or carbonate base.3b In 2002 Peña et al. improved the synthesis of oSAT 5 through the initial silylation of 2-bromophenol 1 with hexamethyldisilazane (HMDS) (Scheme 1a).4 Subsequent halogen-metal exchange then triggered instantaneous retro-Brook O- to C-silyl transfer (3 to 4) prior to triflation.
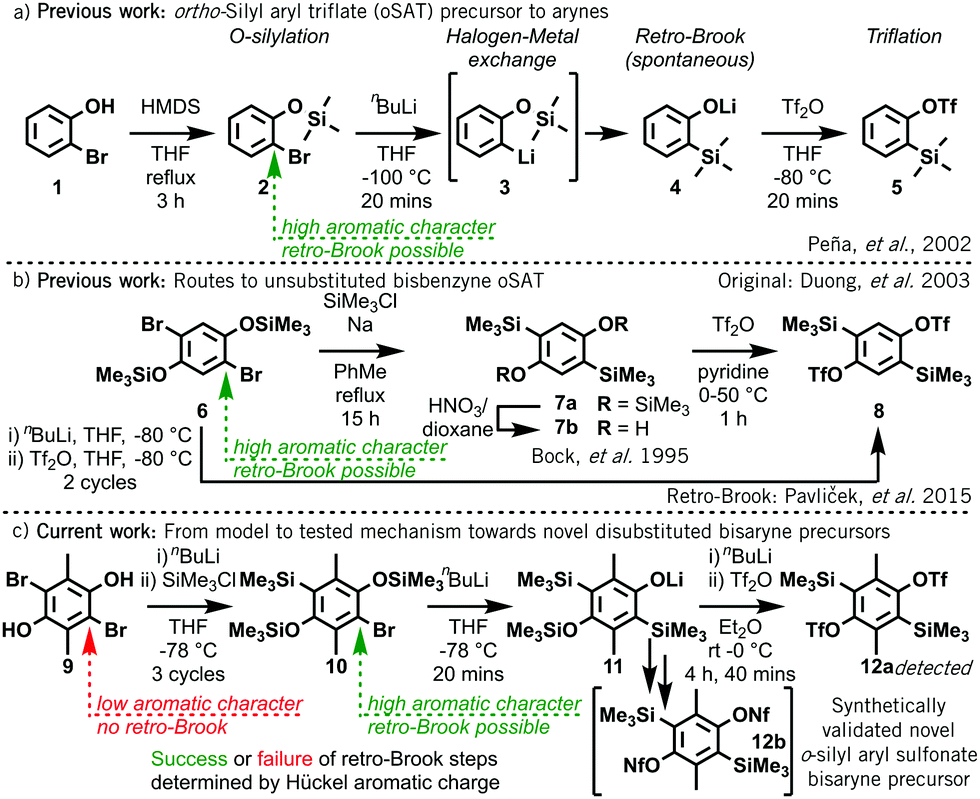 | ||
| Scheme 1 (a) Synthesis of oSAT benzyne precursor 5 from 2-bromophenol 1.4 (b) Approaches to unsubstituted bisbenzyne oSAT precursor 85a–c (c) This work: novel route towards challenging disubstituted bisbenzyne precursors based on simple Hückel aromaticity models. | ||
Whilst this synthetic strategy permits access to many aryne and heteroaryne precursors6 with wide functional group tolerance, it is not universal. In some instances, longer routes are needed, such as Kobayashi's original approach3avia C,O-disilylation, O-desilylation, then triflation.7 This method was used by Duong in 2003 to first produce 1,4-bisbenzyne oSAT precursor 85b before the successful retro-Brook optimization by Pavliček et al. in 2015 (Scheme 1b).5c 1,4-Bisbenzynes from 8 have appeared in around 15 papers,5,8 as well as a recent trisaryne oSAT from its triphenylene trimer,5d yet no additionally substituted examples of oSAT bisbenzynes have been developed.
The different methods of formation of bis/trisarynes have been reviewed by Shi, et al. and usually require more forceful conditions than oSATs, thus limiting functional group tolerance.1d This presents unrealized potential for the rapid generation of functionalized PAHs from bisbenzyne precursors.
In this work, we demonstrate that an electronic barrier is responsible for the failure of the retro-Brook step during the formation of certain oSATs and that a fast and accessible Hückel calculation provides a reliable indication of success. We then validate this model by experimentally confirming three predicted outcomes: (1) a successful new approach to a known benzyne oSAT precursor; (2) the failure of the key step towards a 9,10-phenanthryne oSAT precursor;9 and (3) the development of a synthetic route to a novel difunctionalized bisaryne precursor 12, wherein only one of the two C-silyation steps proceeds via a retro-Brook rearrangement (Scheme 1c).
We hypothesized that o-bromophenyl silyl ethers, such as 2, should undergo halogen-metal exchange then retro-Brook rearrangement if an aromatic charge is present at the brominated position. To this end, Hückel aromatic charges were calculated on MM2 energy-minimized models of a wide range of bromoaryl silyl ether intermediates (2 and 13a–q) towards reported or potential oSAT aryne precursors (Fig. 1).10
 | ||
| Fig. 1 Calculated Hückel aromatic charges at brominated positions in halogen-metal exchange intermediates for the synthesis of a range of aryne precursors (MM2 in PerkinElmer Chem3D).10 | ||
As a benchmark, the intermediate (2) towards unsubstituted benzyne oSAT 5 has an aromatic charge of +0.056 (ESI,† S2).3a The sign of the charge was not important, as 1,3-benzodioxole derivative 13a, reported to undergo successful retro-Brook reaction,6a was calculated at −0.029. A range of literature examples 13c–13m7,11a–i were found to possess charges comparable to benchmark 2, with just 4,5-difluoro 13e11a showing a significantly lower value (+0.005). In light of this difference, it is noteworthy that several mechanisms for halogen-metal exchange have been proposed.12 The model predicted that 3,6-dimethoxy 13n (−0.011) should also undergo retro-Brook rearrangement to the known aryne precursor 13z, originally accessed via C-deprotonation with nBuLi and direct C-silylation of the 2,5-dimethoxyphenol.11j Pleasingly, this novel retro-Brook application proved successful (70% yield; ESI,† S3).
We next considered the extended aromatic precursors 13o–q, with naphthalene derivative 13o11k found to possess a high aromatic charge (+0.076). Interestingly, [5]helicene 13p11l also successfully undergoes retro-Brook rearrangement but displays a significantly lower charge (+0.003). As Hückel approximations presume a planar aromatic system, twisted systems such as 13p appear to represent a limitation of our model. By contrast, the planar 9,10-phenanthryne precursor intermediate 13q has a near-zero aromatic charge (+0.001), which suggests the retro-Brook reaction to be disfavoured;9 indeed, the synthesis of the corresponding phenanthryne oSAT precursor described by Peña et al. in 2000 did not involve this transformation.13a According to Clar's Rule,14 the two peripheral rings in phenanthrene are more aromatic than the central ring, allowing the 9,10-bond to be functionalized in a manner similar to non-aromatic double bonds.15 Even when conjugated alkenes have been exposed to conditions analogous to the retro-Brook reaction, halogen retention was preferred over halogen-metal exchange.16
With a view to accessing novel functionalized bisbenzyne oSATs we decided to apply our Hückel model to test the viability of a retro-Brook-based synthetic approach towards para-dimethylated bisxylyne oSAT 12. As only small positive inductive effects are present, the steric encumbrance common to all pendant substituents can be tested for, while minimizing electronic effects specific to each. Investigations started by modelling the unfunctionalized bisbenzyne oSAT precursor 7 that was accessed via retro-Brook rearrangement by Pavliček et al.5c The MM2 energy-minimized models of post-O-silylation intermediates 13r and 13s confirmed that aromatic character was maintained in both brominated positions (Fig. 2). Although calculations on the xylyne precursor 13t17 revealed suitable aromatic character (+0.026), significant deactivation was found for O-silylated bisxylyne prototype intermediates 13u and 13v. Intriguingly, if the first silyl transfer were to be forced, or silylation achieved via other means, then potential intermediates 13w and 10 for the second stage both have a significant Hückel charge (+0.032). This would suggest that the second silyl transfer should be possible by retro-Brook. Elsewhere, sizeable aromatic charges were calculated for 1,3-diyne precursors 13x (−0.019 and +0.015) and 13y (−0.023), in agreement with the reported retro-Brook syntheses.11m,n
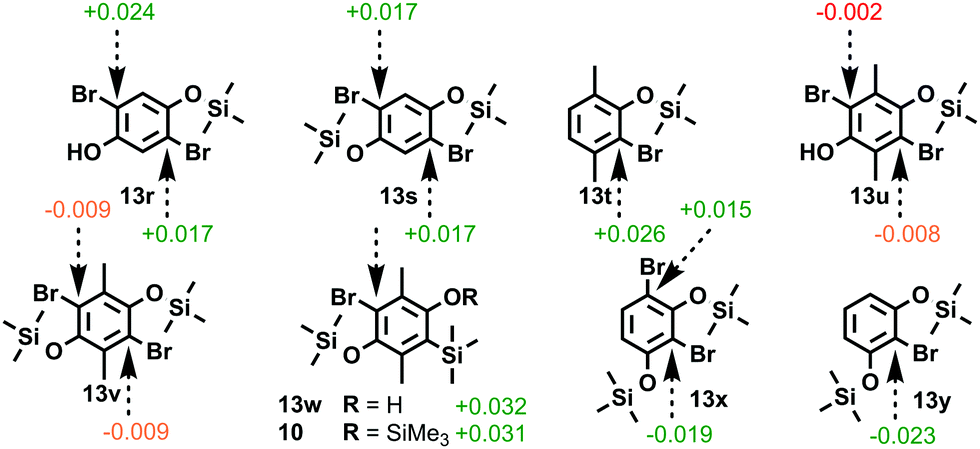 | ||
| Fig. 2 Hückel aromatic charges on brominated positions in halogen-metal exchange intermediates for a range of literature bisbenzyne precursors and prototype models. (MM2 in PerkinElmer Chem3D)10 | ||
Encouraged by the ability of the Hückel model to predict successful retro-Brook syntheses, we experimentally investigated the two outliers: 9,10-phenanthryne and bisxylyne. The phenanthryne precursor 15 was first reported by Peña et al., starting from 9-bromo-10-phenanthrol 14 and prepared using a C,O-disilylation method.13a We successfully repeated this on a gram scale, using an in situ solvent-exchange step to remove impurities and increase the yield by 8% (Scheme 2a).13
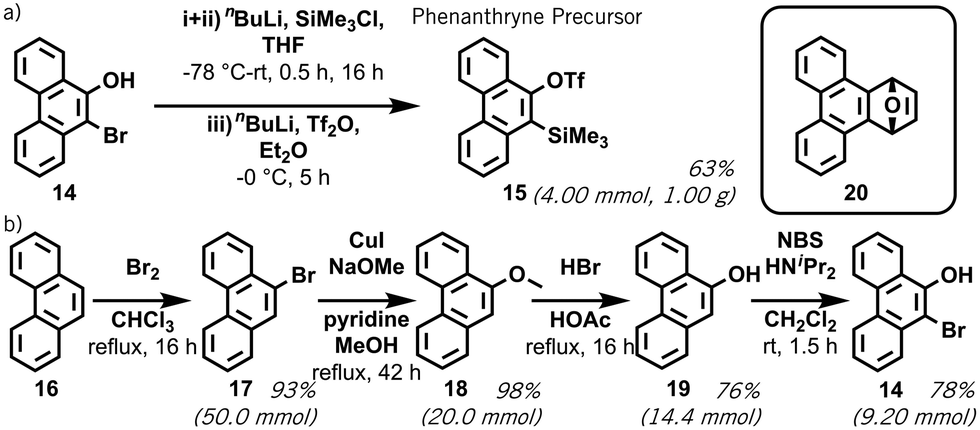 | ||
| Scheme 2 Synthesis of 9,10-phenanthryne precursor 15: our reported improvements in yield and scale for (a) the final step; (b) the conversion of phenanthrene to precursor starting material 9-bromo-10-phenanthrol 14. This does not involve a retro-Brook step. | ||
To test our aromaticity model, attempts were made to form oSAT 15 from 14 using HMDS and a retro-Brook reaction, which – although reported once4 – failed in line with our predictions (ESI,† S5).9 To provide sufficient material for these tests, an improved synthesis of 14 from phenanthrene 16 was developed (Scheme 2b). Interestingly, the bromination of 16 at C-9 and C-10 would not usually be possible at an aromatic carbon without Lewis acid catalyst.15 Our updated synthesis of 15 has: (a) full contemporary purification and characterization; (b) replaces toxic CCl4 with CHCl3; (c) a 76% yield of 19 from 18 (cf. 48%); and (d) a 34% yield of 15 at gram scale over five steps from 16 (ESI,† S4).13a,18 Finally, a quantitative Diels–Alder reaction between 15 and furan was successfully repeated to form adduct 20 (Scheme 2 inset), validating its suitability as an aryne precursor (ESI,† S5).19
Attention now turned to the synthesis of 1,4-bisxylyne oSAT prototype 12a. Our model suggests that the first prospective O- to C-silyl transfer in intermediate 13v is disfavoured, whereas a second C-silyl group could be installed via a retro-Brook reaction using intermediate 13w or 10. To probe this prediction, 2,5-dimethylbenzoquinone 21 was reductively brominated to form 2,5-dibromo-3,6-dimethylhydroquinone 9 (Scheme 3a; ESI,† S6). Attempts to produce bisxylyne oSAT prototype 12a directly from 9 and HMDS via a retro-Brook reaction afforded no product, only crude mixtures highly prone to oxidation (ESI,† S7). Even when forcing conditions were used to access O-silyl intermediate 13v, subsequent sodiation5a to initiate retro-Brook silyl transfer gave an insoluble mixture with no target.
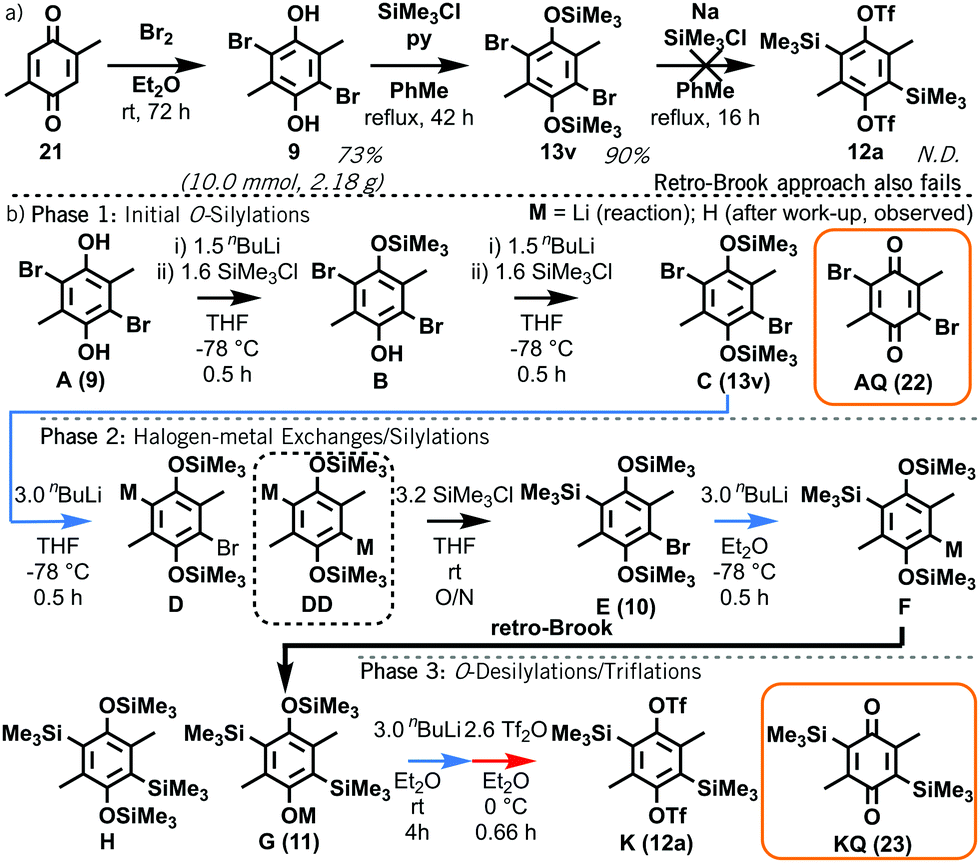 | ||
| Scheme 3 (a) Reductive bromination of 2,5-dimethyl-1,4-benzoquinone 21 with subsequent failed attempts to form a bisxylyne oSAT prototype 12a; (b) abridged putative mechanism of the formation of 12a (K) from 9 (A). Full scheme in ESI,† S9. | ||
Having verified the fates of intermediates 9 and 13v, a synthetic route towards 12a was developed based on our model. Initial O,O′-disilylation and C-silylation of 9, as for phenanthryne oSAT 15, was proposed to access intermediate aryl silane 10. Subsequent retro-Brook reaction and triflation would lead to oSAT 12a (Scheme 3b; ESI,† S8–9). Reaction aliquots were analysed at each stage by NMR (ESI,† S14) and GC/MS (ESI,† S18). The O-silylations (Phase 1) succeeded, although disilylated 13v (C) was observed after the first step and C-lithiated D from halogen-metal exchange after the second, due to excess nBuLi.
Crucially, no retro-Brook reactions were observed with D despite the excess lithiation, as confirmed by a separate test on isolated 13v (ESI,† S10). Following the first C-silylation step (Phase 2), C,C′-dilithiated DD was present despite overnight stirring at room temperature with chlorosilane. This suggests a significant barrier to intermolecular C-silylation ortho to O-silylated species precluding a first retro-Brook (Fig. 3, top). Phenolic signals in the 1H NMR spectrum confirm that the large triply-silylated species must be C,C′,O-isomer G (11), rather than F. Hence, a retro-Brook reaction on the second side is successful, in line with our mechanistic model. After solvent exchange and further nBuLi, without further silylation, the next aliquot contains only G (11), F and tetrasilylated H (Fig. 3, middle). Here, the 30 minute reaction time allows E (10) to undergo halogen-metal exchange to afford F but retro-Brook to G (11) cannot complete; H can only form from O-silyl exchanges. Unfortunately, after triflation and work-up (Phase 3), quinone KQ (23) predominated (Fig. 3, bottom, 41% isolated yield); despite this, C,C′-disilylated K (12a) is detected in situ and the oxidation product successfully confirms our assignments. H was later isolated in 74% yield but attempts to introduce the triflate only produced KQ (23, 56% yield). This sensitivity of the triflates suggests this may be an inherent limitation of functionalized bis-oSAT precursors. However, transient o-silylaryl nonaflates such as 12b (putative structure, accessed from air-stable H) are also aryne precursors9 and pleasingly the subsequent Diels–Alder reaction of furan with the bisaryne proceeded successfully in 13% yield (Scheme 4).
 | ||
| Fig. 3 Partial stack plot of GC/MS aliquots from the formation of bisxylyne oSAT prototype 12a (K). Letters correspond to Scheme 3b. | ||
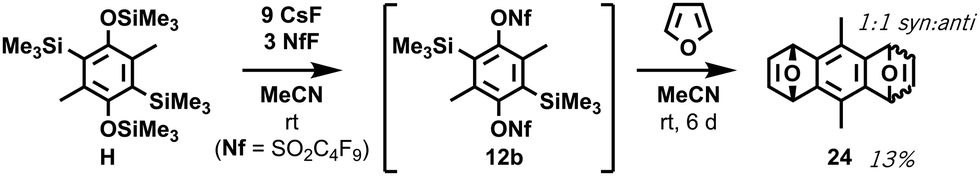 | ||
| Scheme 4 Successful bisxylene formation from transient ortho-silylaryl nonaflate 12b to form adduct 24 by in situ Diels–Alder reaction. See ESI,† S12. | ||
In summary, we have developed a quick and facile MM2/Hückel modelling strategy to test the success of retro-Brook O- to C-silyl transfer relevant to aryne precursor synthesis. A novel application (13z) and an expected failure (15) were validated experimentally. Next, a route towards 1,4-bisxylyne precursors 12a/b was developed with a retro-Brook step for just one C-silylation. This is the first C,C′-disubstituted o-silylaryl sulfonate bisbenzyne precursor: with appropriate substituents, many previously elusive bisaryne precursors may now be accessible.
We are grateful to the EPSRC (EP/M026221/1, CRJ) and QMUL (teaching laboratory technician/teaching fellow posts for EAN; studentship to AYRW) for financial support. We thank the National Mass Spectrometry Facility at Swansea University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) H. H. Wenk, M. Winkler and W. Sander, Angew. Chem., Int. Ed., 2003, 42, 502–528 CrossRef CAS; (b) D. Pérez, D. Peña and E. Guitián, Chem. – Eur. J., 2013, 5981–6103 Search PubMed; (c) S. S. Bhojgude, A. Bhunia and A. T. Biju, Acc. Chem. Res., 2016, 49, 1658–1670 CrossRef CAS; (d) J. Shi, Y. Li and Y. Li, Chem. Soc. Rev., 2017, 46, 1707–1719 RSC; (e) T. Roy and A. T. Biju, Chem. Commun., 2018, 54, 2580–2594 RSC; (f) D. B. Werz and A. T. Biju, Angew. Chem., Int. Ed., 2019, 59, 3385–3398 CrossRef.
- Recent cases: (a) A. V. Kelleghan, C. A. Busacca, M. Sarvestani, I. Volchkov, J. M. Medina and N. K. Garg, Org. Lett., 2020, 22, 1665–1669 CrossRef CAS; (b) J. Xu, S. Li, H. Wang, W. Xu and S. Tian, Chem. Commun., 2017, 53, 1708–1711 RSC; (c) R. Fan, B. Liu, T. Zheng, K. Xu, C. Tan, T. Zeng, S. Su and J. Tan, Chem. Commun., 2018, 54, 7081–7084 RSC; (d) H. Takikawa, A. Nishii, H. Takiguchi, H. Yagishita, M. Tanaka, K. Hirano, M. Uchiyama, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2020, 59, 12440–12444 CrossRef CAS.
- (a) Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211–1214 CrossRef CAS; (b) S. Yoshida, Y. Hazama, Y. Sumida, T. Yano and T. Hosoya, Molecules, 2015, 20, 10131–10140 CrossRef CAS.
- D. Peña, A. Cobas, D. Pérez and E. Guitián, Synthesis, 2002, 1454–1458 Search PubMed.
- (a) H. Bock, S. Nick, C. Näther and K. Ruppert, Z. Naturforsch., 1995, 50, 595–604 CAS; (b) H. M. Duong, M. Bendikov, D. Steiger, Q. Zhang, G. Sonmez, J. Yamada and F. Wudl, Org. Lett., 2003, 5, 4433–4436 CrossRef CAS; (c) N. Pavliček, B. Schuler, S. Collazos, N. Moll, D. Pérez, E. Guitián, G. Meyer, D. Peña and L. Gross, Nat. Chem., 2015, 7, 623–628 CrossRef; (d) I. Pozo, D. Peña, E. Guitían and D. Pérez, Chem. Commun., 2020, 56, 12853–12856 RSC.
- (a) Y. Sato, T. Tamura and M. Mori, Angew. Chem., Int. Ed., 2004, 43, 2436–2440 CrossRef CAS; (b) F. I. M. Idiris, C. E. Majesté, G. B. Craven and C. R. Jones, Chem. Sci., 2018, 9, 2873–2878 RSC.
- A. B. Smith and W.-S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6787–6792 CrossRef CAS.
- Selected examples: (a) R. Shintani, H. Otomo, K. Ota and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 7305–7308 CrossRef CAS; (b) K. G. U. R. Kumarasinghe, F. R. Fronczek, H. U. Valle and A. Sygula, Org. Lett., 2016, 18, 3054–3057 CrossRef CAS; (c) P. Asgari, U. S. Dakarapu, H. H. Nguyen and J. Jeon, Tetrahedron, 2017, 73, 4052–4061 CrossRef CAS; (d) X. Yang and G. C. Tsui, Chem. Sci., 2018, 9, 8871–8875 RSC; (e) W. Xu, X.-D. Yang, X.-B. Fan, X. Wang, C.-H. Tung, L.-Z. Wu and H. Cong, Angew. Chem., Int. Ed., 2019, 58, 3943–3947 CrossRef CAS; (f) Most recent of seven works: I. Pozo, Z. Majzik, N. Pavliček, M. Melle-Franco, E. Guitián, D. Peña, L. Gross and D. Pérez, J. Am. Chem. Soc., 2019, 141, 15488–15493 CrossRef CAS.
- T. Ikawa, S. Masuda, H. Nakajima and S. Akai, J. Org. Chem., 2017, 82, 4242–4253 CrossRef CAS . See ESI† S5.
- ChemOffice Professional, PerkinElmer Informatics, 2019.
- (a) V. Singh, R. S. Verma, A. K. Khatana and B. Tiwari, J. Org. Chem., 2019, 84, 14161–14167 CrossRef CAS; (b) Y. Tsuchido, T. Ide, Y. Suzaki and K. Osakada, Bull. Chem. Soc. Jpn., 2015, 88, 821–823 CrossRef CAS; (c) H. Jiang, Y. Zhang, W. Xiong, J. Cen, L. Wang, R. Cheng, C. Qi and W. Wu, Org. Lett., 2019, 21, 345–349 CrossRef CAS; (d) C.-H. Sun, Y. Lu, Q. Zhang, R. Lu, L.-Q. Bao, M.-H. Shen and H.-D. Xu, Org. Biomol. Chem., 2017, 15, 4058–4063 RSC; (e) K. Nogi, T. Fujihara, J. Terao and Y. Tsuji, J. Org. Chem., 2015, 80, 11618–11623 CrossRef CAS; (f) C. Hall, J. L. Henderson, G. Ernouf and M. F. Greaney, Chem. Commun., 2013, 49, 7602 RSC; (g) Y. Zeng, L. Zhang, Y. Zhao, C. Ni, J. Zhao and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955–2958 CrossRef CAS; (h) J. Pan, M. Su and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 8647–8651 CrossRef CAS; (i) B. Lakshmi, U. Wefelscheid and U. Kazmaier, Synlett, 2011, 345–348 CAS; (j) J. George, J. S. Ward and M. S. Sherburn, Org. Lett., 2019, 21, 7529–7533 CrossRef CAS; (k) T.-Y. Zhang, C. Liu, C. Chen, J.-X. Liu, H.-Y. Xiang, W. Jiang, T.-M. Ding and S.-Y. Zhang, Org. Lett., 2018, 20, 220–223 CrossRef CAS; (l) T. Hosokawa, Y. Takahashi, T. Matsushima, S. Watanabe, S. Kikkawa, I. Azumaya, A. Tsurusaki and K. Kamikawa, J. Am. Chem. Soc., 2017, 139, 18512–18521 CrossRef CAS; (m) T. Ikawa, S. Masuda, A. Takagi and S. Akai, Chem. Sci., 2016, 7, 5206–5211 RSC; (n) J. Shi, D. Qiu, J. Wang, H. Xu and Y. Li, J. Am. Chem. Soc., 2015, 137, 5670–5673 CrossRef CAS.
- T. D. Sheppard, Org. Biomol. Chem., 2009, 7, 1043–1052 RSC.
- (a) D. Peña, D. Pérez, E. Guitián and L. Castedo, J. Org. Chem., 2000, 65, 6944–6950 CrossRef; (b) M. Idris, C. Coburn, T. Fleetham, J. Milam-Guerrero, P. I. Djurovich, S. R. Forrest and M. E. Thompson, Mater. Horiz., 2019, 6, 1179–1186 RSC.
- E. Clar, Polycyclic Hydrocarbons, Springer, Berlin, 1964 Search PubMed.
- C. A. Dornfeld, J. E. Callen and G. H. Coleman, Org. Synth., 1948, 28, 19–21 CrossRef CAS . Bromine adds across the 9,10-bond prior to rearomatization on heating.
- (a) T. J. Barton and B. L. Groh, J. Am. Chem. Soc., 1985, 107, 7221–7222 CrossRef CAS; (b) N. Shimizu, F. Shibata and Y. Tsuno, Bull. Chem. Soc. Jpn., 1987, 60, 777–778 CrossRef CAS; (c) L. Duhamel, J. Gralak and B. Ngono, J. Organomet. Chem., 1989, 363, C4–C6 CrossRef CAS; (d) L. Duhamel, J. Gralak and A. Bouyanzer, Tetrahedron Lett., 1993, 34, 7745–7748 CrossRef CAS; (e) L. Duhamel, J. Gralak and B. Ngono, J. Organomet. Chem., 1994, 464, C11–C13 CrossRef CAS.
- Y. Wang, A. D. Stretton, M. C. McConnell, P. A. Wood, S. Parsons, J. B. Henry, A. R. Mount and T. H. Galow, J. Am. Chem. Soc., 2007, 129, 13193–13200 CrossRef CAS.
- (a) R. G. R. Bacon and S. C. Rennison, J. Chem. Soc. C, 1969, 312–315 RSC; (b) S. Kobayashi, K. Kitamura, A. Miura, M. Fukuda, M. Kihara and M. Azekawa, Chem. Pharm. Bull., 1972, 20, 694–699 CrossRef CAS.
- M. McKee, J. Haner, E. Carlson and W. Tam, Synthesis, 2014, 1518–1524 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc08283j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |