
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultivalent Sgc8c-aptamer decorated polymer scaffolds for leukemia targeting†
Zhaobao
Zhang
 a,
Chunling
Tang
b,
Roel
Hammink
cd,
Frank H. T.
Nelissen
a,
Hans A.
Heus
*a and
Paul H. J.
Kouwer
*a
a,
Chunling
Tang
b,
Roel
Hammink
cd,
Frank H. T.
Nelissen
a,
Hans A.
Heus
*a and
Paul H. J.
Kouwer
*a
aInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: p.kouwer@science.ru.nl
bRadiotherapy & OncoImmunology Laboratory, Department of Radiation Oncology, Radboud Institute for Molecular Life Sciences, Radboud University Medical Centre, Geert Grooteplein 26, 6525 GA Nijmegen, The Netherlands
cDepartment of Tumor Immunology, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Geert Grooteplein 26, 6525 GA Nijmegen, The Netherlands
dDivision of Immunotherapy, Oncode Institute, Radboud University Medical Center, Nijmegen, The Netherlands
First published on 4th February 2021
Abstract
T-cell acute lymphoblastic leukemia causes a disproportional amount of immature white blood cells in the patients’ bone marrow. The significant undesired side effects associated with traditional chemotherapy treatment prompted us to study a more effective treatment strategy. We decorated polyisocyanopeptide scaffolds with the selective leukemia cell binding aptamer sgc8c and found that the polymers inhibit proliferation by G0/G1-phase arrest, serving as an opportunity for future therapeutic strategies.
T-cell acute lymphoblastic leukemia (T-ALL) is a malignant disease that alters cell proliferation, survival and maturation, and eventually leads to a lethal accumulation of leukemia cells that outcompete normal white blood cells in the patients’ bone marrow.1 It has a high survival rate in childhood2 but the cure rate of adults rarely exceeds 40%.3 The most widely used treatment is traditional chemotherapy, targeting fast-growing cells, both cancerous and healthy, which inevitably leads to side-effects, such as immunosuppression, anemia and osteonecrosis. It is essential to develop new targeted therapies for T-ALL, with minimized interactions with host factors, improved efficacy and reduced toxic effects.
Aptamers are single-stranded DNA or RNA oligonucleotides that are developed to bind to specific targets with high specificity and affinity.4–6 Recent work highlighted aptamers that inhibit virus infections7 and modify T-cell responses,8 demonstrating their great promise for nucleic acid-based therapeutics. The DNA aptamer sgc8c has been proposed to target T-ALL as it directly binds the transmembrane protein tyrosine kinase-7 (PTK7), which is highly expressed in many leukemic and other cancerous cell lines.9–11 Besides a high specificity and affinity for PTK7 (Kd = 0.8 nM),5,12 sgc8c offers the advantage of being a small and easy to synthesize molecule.13,14 New anti-T-ALL therapeutic tools include sgc8c-conjugated chemotherapy drugs,15,16 and sgc8c-decorated platinum17 and gold nanoparticles.18 The potential disadvantages of these strategies include cellular toxicity to neighboring tissue after aptamer release.
Mechanistically, sgc8c binds PTK7 that regulates the Wnt/β-catenin signaling pathway,19,20 a key pathway in regulating cell proliferation and tumorgenesis. Knockdown of PTK7 inhibits Wnt pathway activation, while overexpression leads to increased metastatic events.19,21–23 After activation PTK7 is internalized into the cytoplasm.24–26 In analogy to the work from Groves and coworkers,27 we hypothesize that restricting PTK7 movement with a potent multivalent target will lead to inhibition of its downstream signaling in T-ALL.
In this study, we grafted sgc8c to a high molecular weight synthetic semi-flexible polymer, based on an oligo(ethylene glycol)-decorated polyisocyanopeptide (PIC), which was recently developed by our group.28,29 PICs have been developed into promising materials for cell culture applications,30–32 wound healing,33 and as a scaffold for immunological therapies.34 Earlier studies demonstrated that PICs are fully biocompatible.33 Using the relatively rigid PIC polymers as a scaffold, we anticipate that the multivalent sgc8c aptamer presentation induces a far more effective response at low aptamer concentrations.34–36 In addition, conjugation to a high molecular weight polymer backbone prevents rapid clearance of aptamers by renal filtration and enhances nuclease resistance once applied in vivo.13,37 We show that aptamer-decorated PIC efficiently targets leukemia cells and inhibits their growth by inducing G0/G1 arrest. As such, the material holds great promise for targeted T-ALL therapies, aiming to minimize adverse effects to healthy tissue and, therefore, side effects.
Polymer synthesis and functionalization: The materials were prepared using well-established protocols.38 In brief, azide-appended PIC (PIC-N3) was prepared by copolymerization of the methoxy-tetra(ethylene glycol) monomer (96.7%) and the azide-functionalized monomer (3.3%, Fig. 1), yielding high molecular weight polymers with Mn = 1150 kDa as measured by AFM (Fig. S1, ESI†). Based on the feed ratio of both monomers, on average 97 azide groups are displayed along a single polymer. Synthesis details and polymer characterization39 (Fig. S2 and S3) are provided in the ESI.†
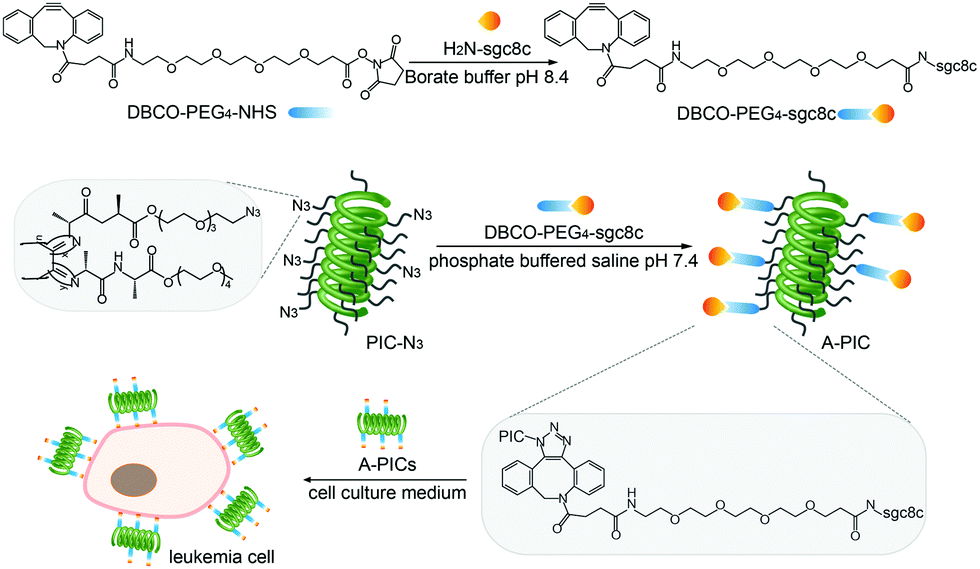 | ||
| Fig. 1 Structure and preparation of sgc8c-decorated PIC (A-PIC). Note that adjacent aptamers are statistically spaced ∼3.5 nm. Details are given in the ESI.† | ||
In parallel, the amine terminus of sgc8c (17 nucleotides) as well as its fluorescently-labeled equivalent were equipped with a reactive cyclooctyne (DBCO) group through standard NHS chemistry. The reaction of the (fluorescent) DBCO-aptamers to the azide moieties on the PIC polymer backbone yielded the desired sgc8c aptamer-decorated polymers (A-PIC). Using gel electrophoresis, supported by a fluorescent dye assay for free DBCO (Fig. S4, ESI†), we determined a degree of conjugation of ∼90%, which means that, on average, nearly 90 aptamers are displayed on a single PIC chain.35,38 Nuclease stability of A-PIC is confirmed after incubation of the polymer in phosphate buffered saline (PBS) with fetal calf serum (FCS, 10%) over 24 h (Fig. S5, ESI†).37
A-PIC efficiently binds leukemia cells: CCRF-CEM leukemia cells express the transmembrane protein PTK7, to which the sgc8c aptamer binds.5,12 To confirm that A-PICs target PTK7 on the cell surface of leukemia cells, we incubated the cells with fluorescently labeled sgc8c (6-FAM-A-PIC). As a control, we also incubated the cells with the free aptamer with the same fluorescent label (Fig. 2). After 2 h, we clearly observed fluorescence signals from the cell membranes in both experiments, indicating efficient targeting. The fluorescence from the A-PIC group, however, is remarkably stronger. Quantitative analysis shows that binding of A-PICs to CCRF-CEM leukemia cells is highly efficient, reaching up to 75% (in 10 different fields of observation) whereas free sgc8c only binds to 40% of cells (Fig. 2C). In addition, we underline the much higher fluorescence intensity of the cells incubated with the A-PIC, which indicates that much more aptamer is bound.
 | ||
| Fig. 2 A-PIC targets CCRF-CEM leukemia cells better than free sgc8c. (A and B) Microscopy analysis (I bright field, II fluorescence and III merge) showing CCRF-CEM bound with (A) fluorescently labeled aptamer (6-FAM-sgc8c) and (B) fluorescently labeled A-PIC (6-FAM-A-PIC) at the same aptamer concentration (1.25 μM). The data show binding after 2 h of incubation with the same microscope settings. Scale bar 25 μm. (C) Quantification of fluorescence-positive cells in 3 (free sgc8c) or 10 (A-PIC) different fields of observation. Data shows mean and standard deviation. Statistics: ***, p < 0.001, by Student's t-test. | ||
A-PIC inhibits leukemia cell proliferation. We then investigated the effect of A-PIC binding to CCRF-CEM leukemia cells on their proliferation. Following our hypothesis, a stronger inactivation of PTK7 by A-PICs is anticipated to give rise to reduced proliferation and, ultimately, to reduced cancer progress. We studied the dose-dependent inhibition in a series of different A-PICs/PIC-N3 ratios designed to vary the aptamer concentration (100%, 40%, 10% A-PIC or 1.25, 0.5, 0.125 μM, respectively) while keeping the PIC concentration constant (2 mg mL−1, Fig. 3). As controls, we included a sample with free sgc8c (1.25 μM, which is the same concentration as 100% A-PIC), a PIC-N3 only sample (2 mg mL−1) and culture medium only.
 | ||
| Fig. 3 CCRF-CEM leukemia cell proliferation of three treatment groups (10% A-PICs, 40% A-PICs and 100% A-PICs), and three control groups (PBS buffer, PIC-N3 only and free sgc8c). Cell numbers were counted with a Bio-Rad TC20™ automated cell counter after mixing with Trypan blue. Data are collected from five independent experiments on different well plates. The data are shown as mean ± standard deviation. Statistics: **, p < 0.01, ***, p < 0.001, n = 5, tested by one-way ANOVA followed by a Tukey's multiple comparison test. | ||
After 2 days of incubation in the appropriate conditions, we observed the strongest inhibition of leukemia cell growth in the 100% A-PICs sample (Fig. 3). In comparison with the control that was treated with PBS buffer only, we observe a more than one-third reduction in cell number (4.2 × 105vs. 6.6 × 105, p < 0.001) for the 100% A-PICs group. Partially functionalized A-PIC mixtures (40% A-PICs group, 5.1 × 105vs. 6.6 × 105, p < 0.01) showed less pronounced inhibition on cell proliferation and we find no significant difference between the 10% A-PICs group and the PBS control group. Note that the PIC polymer in itself has no effect on cell proliferation (PIC-N3vs. PBS, p = 0.70), which is consistent with earlier work and confirms that PIC is a biocompatible material that displays no significant cell toxicity.33
We also compared different A-PIC mixtures (10%, 40%, and 100% A-PICs groups) to the free sgc8c group as we hypothesized that through multivalency, the PIC scaffold enhances the efficiency of conjugated sgs8c aptamer. Indeed, compared to free sgc8c at the same aptamer concentration, the 100% A-PIC group shows a significant reduction in cell density after 2 days of incubation (4.2 × 105vs. 5.8 × 105, p < 0.01). Reducing the sgc8c density 2.5-fold on the PIC (40% PIC) gives equal or stronger suppression of proliferation than the free aptamer, highlighting the benefits of the multivalency approach. Comparison between the different aptamer-decorated polymers only gives a significant difference between the 10 and 100% A-PIC groups (4.2 × 105vs. 5.8 × 105, p < 0.01).
A-PIC inhibits leukemia growth by G0-phase arrest: To better characterize the inhibition mechanism of leukemia cell growth from the cell cycle perspective, we performed fluorescence-activated cell sorting (FACS). We selected the 100% A-PIC and the PBS control, which showed the strongest difference in proliferative activity. Cells were incubated with either PBS or 100% A-PICs and, after 2 days, stained with propidium iodide and analyzed by FACS. The PBS control group provides a baseline distribution with cell numbers in each phase (Fig. 4A and Fig. S4, ESI†). After two days of incubation, leukemia cells treated with A-PIC showed an increase (56.5% vs. 49.7%) in the number of cells in G0/G1 phase and a concomitant decrease (14.3% vs. 19.9%) in G2/M phase (Fig. 4B and ESI,† Fig. S2), compared to the PBS control. The results suggest a G0/G1-phase arrest. FACS analysis from four independent experiments revealed a small but statistically significant increase in the fraction of cells in the G0/G1 phase (7.4% increase, p < 0.01; Fig. 4C) accompanied by a decrease of cells in the G2/M phase (6.4% decrease, p < 0.01; Fig. 4C). The observed G0/G1-phase arrest in targeted leukemia cells likely leads to a delayed re-entering into the cell division cycle, the G2/M phase, which stochastically controls the number of final cell divisions and, thus, eventually results in a decreased proliferation.
 | ||
| Fig. 4 Cell cycle analysis. (A and B) Flow cytometry histograms of the cell cycle analysis of CCRF-CEM leukemia cells after 2 days of incubation with PBS (A) or 100% A-PICs (B) showing one representative result of four independent experiments; the complete set of original data is shown in Fig. S6 and S7 (ESI†). The left bivariate histogram of forward scatter (FSC)/side scatter (SSC) analysis illustrates the gating strategy to eliminate debris and dead cells. The right panel shows the cell density percentages for the G0/G1, S and G2/M phases, defined by their DNA content measured by propidium iodide. (C) Cell cycle changes with A-PICs treatment averaged for four experiments, shown as average ± standard deviation. **, p < 0.01, n = 4, tested by two-way ANOVA. | ||
Discussion: Previously, our group has decorated PIC polymers with antibodies against CD3 and CD28 to generate synthetic dendritic cells for modulating T-cell responses.34,35 The excellent presentation of factors along an extended semi-flexible backbone like that of PIC offers a great advantage compared to fully flexible polymers or fully rigid nanoparticles.40
In this work, we provide a novel small molecule alternative approach using aptamers. Aptamers hold great potential with many favorable characteristics, such as their small size, high stability, low immunogenicity and facile chemical synthesis.
Nucleic acid-based therapeutics are quickly emerging as a strong alternative or co-therapy to chemical agents.7 In addition, highly specific aptamer-functionalized agents are extensively studied for their use in targeted therapies.41 The sgc8c grafted to the PIC scaffold binds to PTK7 and effectively reduces leukemic cell proliferation by prolonging the G0/G1 phase. The limited effect of soluble PTK7 underlines the benefits brought forward by the multivalent presentation of aptamers on the polymer backbone. Future work has to unveil what the optimal aptamer density along the polymer backbone is and how treatments should be optimized to further suppress proliferation.
PTKs play a key role in growth factor-related signal modulation and apoptosis.42–45 PTK knockdown experiments revealed Wnt pathway deactivation and delayed entry towards the G2/M phase, in line with our results. From this initial work it is hard to delineate how and to which extent PTK7 activity is reduced by A-PICs, however, future gene expression profiling by RNA-sequencing may help to elucidate the underlying mechanism.
Beyond sgc8c in the leukemia treatment context, PICs are becoming increasingly attractive as a biofactor-presenting scaffold, both in the soluble form and as a gel. As conjugation strategies have been well-described, the opportunities to present one or multiple bioactive factors selectively to cells are virtually endless.
In summary, with our novel A-PIC design, we observed a reduction of leukemia cell proliferation already after 2 days, which makes the polymer hybrid a promising material for targeted anticancer treatment with reduced side effects. Having demonstrated the potency of our A-PICs, our next goal is to optimize the polymers and to test A-PICs with in vivo mouse models. In addition, we aim to optimize A-PICs for solid tumor therapy as the PIC polymers are readily transformed into a locally administrable gel-like ECM material.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- C.-H. Pui, in Encyclopedia of Cancer, ed. M. Schwab, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 23–26 DOI:10.1007/978-3-642-16483-5_57.
- A. Look, Lancet, 2008, 371, 1030–1043 CrossRef.
- R. Bassan and D. Hoelzer, J. Clin. Oncol., 2011, 29, 532–543 CrossRef.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS.
- D. Shangguan, Y. Li, Z. Tang, Z. C. Cao, H. W. Chen, P. Mallikaratchy, K. Sefah, C. J. Yang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838–11843 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818 CrossRef CAS.
- J. Zhou, S. Satheesan, H. Li, M. S. Weinberg, K. V. Morris, J. C. Burnett and J. J. Rossi, Chem. Biol., 2015, 22, 379–390 CrossRef CAS.
- J. O. McNamara, D. Kolonias, F. Pastor, R. S. Mittler, L. Chen, P. H. Giangrande, B. Sullenger and E. Gilboa, J. Clin. Invest., 2008, 118, 376–386 CrossRef CAS.
- D. Shangguan, Z. Cao, L. Meng, P. Mallikaratchy, K. Sefah, H. Wang, Y. Li and W. Tan, J. Proteome Res., 2008, 7, 2133–2139 CrossRef CAS.
- Z. Xiao, D. Shangguan, Z. Cao, X. Fang and W. Tan, Chem. – Eur. J., 2008, 14, 1769–1775 CrossRef CAS.
- M. Leitner, A. Poturnayova, C. Lamprecht, S. Weich, M. Snejdarkova, I. Karpisova, T. Hianik and A. Ebner, Anal. Bioanal. Chem., 2017, 409, 2767–2776 CrossRef CAS.
- D. Shangguan, Z. Tang, P. Mallikaratchy, Z. Xiao and W. Tan, ChemBioChem, 2007, 8, 603–606 CrossRef CAS.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537 CrossRef CAS.
- T. Hermann and D. J. Patel, Science, 2000, 287, 820–825 CrossRef CAS.
- Y. F. Huang, D. Shangguan, H. Liu, J. A. Phillips, X. Zhang, Y. Chen and W. Tan, ChemBioChem, 2009, 10, 862–868 CrossRef CAS.
- S.-T. Kang, Y.-L. Luo, Y.-F. Huang and C.-K. Yeh, DNA-conjugated gold nanoparticles for ultrasound targeted drug delivery, 2012 IEEE International Ultrasonics Symposium, Dresden, 2012, pp. 1866–1868 Search PubMed.
- N. L. McGinely, J. A. Plumb and N. J. Wheate, J. Inorg. Biochem., 2013, 128, 124–130 CrossRef CAS.
- X. Ye, H. Shi, X. He, Y. Yu, D. He, J. Tang, Y. Lei and K. Wang, Nanoscale, 2016, 8, 2260–2267 RSC.
- N. Bin-Nun, H. Lichtig, A. Malyarova, M. Levy, S. Elias and D. Frank, Development, 2014, 141, 410–421 CrossRef CAS.
- F. Puppo, V. Thomé, A. C. Lhoumeau, M. Cibois, A. Gangar, F. Lembo, E. Belotti, S. Marchetto, P. Lécine and T. Prébet, EMBO Rep., 2011, 12, 43–49 CrossRef CAS.
- A.-C. Lhoumeau, M.-L. Arcangeli, M. De Grandis, M. Giordano, J.-C. Orsoni, F. Lembo, F. Bardin, S. Marchetto, M. Aurrand-Lions and J.-P. Borg, J. Immunol., 2016, 196, 4367–4377 CrossRef CAS.
- A.-C. Lhoumeau, S. Martinez, J.-M. Boher, G. Monges, R. Castellano, A. Goubard, M. Doremus, F. Poizat, B. Lelong and C. de Chaisemartin, PLoS One, 2015, 10 Search PubMed.
- T. Prebet, A.-C. Lhoumeau, C. Arnoulet, A. Aulas, S. Marchetto, S. Audebert, F. Puppo, C. Chabannon, D. Sainty and M.-J. Santoni, Blood, 2010, 116, 2315–2323 CrossRef CAS.
- H.-W. Na, W.-S. Shin, A. Ludwig and S.-T. Lee, J. Biol. Chem., 2012, 287, 25001–25009 CrossRef CAS.
- V. S. Golubkov, A. V. Chekanov, P. Cieplak, A. E. Aleshin, A. V. Chernov, W. Zhu, I. A. Radichev, D. Zhang, P. D. Dong and A. Y. Strongin, J. Biol. Chem., 2010, 285, 35740–35749 CrossRef CAS.
- V. S. Golubkov, N. L. Prigozhina, Y. Zhang, K. Stoletov, J. D. Lewis, P. E. Schwartz, R. M. Hoffman and A. Y. Strongin, J. Biol. Chem., 2014, 289, 24238–24249 CrossRef CAS.
- K. Salaita, P. M. Nair, R. S. Petit, R. M. Neve, D. Das, J. W. Gray and J. T. Groves, Science, 2010, 327, 1380–1385 CrossRef CAS.
- P. H. J. Kouwer, M. Koepf, V. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto and R. Hoogenboom, Nature, 2013, 493, 651–655 CrossRef CAS.
- S. Mandal, Z. H. Eksteen-Akeroyd, M. J. Jacobs, R. Hammink, M. Koepf, A. J. Lambeck, J. C. van Hest, C. J. Wilson, K. Blank and C. G. Figdor, Chem. Sci., 2013, 4, 4168–4174 RSC.
- K. Liu, S. M. Mihaila, A. Rowan, E. Oosterwijk and P. H. J. Kouwer, Biomacromolecules, 2019, 20, 826–834 CrossRef CAS.
- Y. Zhang, C. Tang, P. N. Span, A. E. Rowan, T. W. Aalders, J. A. Schalken, G. J. Adema, P. H. J. Kouwer, M. M. P. Zegers and M. Ansems, Adv. Sci., 2020, 7, 202001797 Search PubMed.
- Y. Zhang, M. M. Zegers, A. E. Rowan, P. N. Span and P. H. J. Kouwer, Adv. Sci., 2021 DOI:10.1002/advs.202003380.
- R. C. Op‘t Veld, O. I. van den Boomen, D. M. Lundvig, E. M. Bronkhorst, P. H. Kouwer, J. A. Jansen, E. Middelkoop, J. W. Von den Hoff, A. E. Rowan and F. A. Wagener, Biomaterials, 2018, 181, 392–401 CrossRef.
- R. Hammink, S. Mandal, L. J. Eggermont, M. Nooteboom, P. H. G. M. Willems, J. Tel, A. E. Rowan, C. G. Figdor and K. G. Blank, ACS Omega, 2017, 2, 937–945 CrossRef CAS.
- L. J. Eggermont, R. Hammink, K. G. Blank, A. E. Rowan, J. Tel and C. G. Figdor, Adv. Ther., 2018, 1, 1800021 CrossRef.
- Y. Song, Y. Shi, M. Huang, W. Wang, Y. Wang, J. Cheng, Z. Lei, Z. Zhu and C. Yang, Angew. Chem., Int. Ed., 2019, 58, 2236–2240 CrossRef CAS.
- L. Yang, M. Liang, C. Cui, X. Li, L. Li, X. Pan, H. Safari Yazd, M. Hong, J. Lu, Y. C. Cao and W. Tan, ChemBioChem, 2021 DOI:10.1002/cbic.202000712.
- S. R. Deshpande, R. Hammink, R. K. Das, F. H. Nelissen, K. G. Blank, A. E. Rowan and H. A. Heus, Adv. Funct. Mater., 2016, 26, 9075–9082 CrossRef CAS.
- M. Koepf, H. J. Kitto, E. Schwartz, P. H. J. Kouwer, R. J. M. Nolte and A. E. Rowan, Eur. Polym. J., 2013, 49, 1510–1522 CrossRef CAS.
- D. Voerman, M. Schluck, J. Weiden, B. Joosten, L. J. Eggermont, T. van den Eijnde, B. Ignacio, A. Cambi, C. G. Figdor, P. H. J. Kouwer, M. Verdoes, R. Hammink and A. E. Rowan, Biomacromolecules, 2019, 20, 2587–2597 CrossRef CAS.
- X. Li, Q. Zhao and L. Qiu, J. Controlled Release, 2013, 171, 152–162 CrossRef CAS.
- T. Burke, Drugs Future, 1992, 17, 119–131 Search PubMed.
- M. D. Hollenberg, Mol. Cell. Biochem., 1995, 149, 77–85 CrossRef.
- E. Kyle, L. Neckers, C. Takimoto, G. Curt and R. Bergan, Mol. Pharmacol., 1997, 51, 193–200 CrossRef CAS.
- F. Lian, M. Bhuiyan, Y. W. Li, N. Wall, M. Kraut and F. H. Sarkar, Nutr. Cancer, 1998, 31, 184–191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc08205h |
| This journal is © The Royal Society of Chemistry 2021 |