
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodaelectro-catalyzed chemo-divergent C–H activations with alkylidenecyclopropanes for selective cyclopropylations†
Zhigao
Shen
,
Isaac
Maksso
,
Rositha
Kuniyil
,
Torben
Rogge
 and
Lutz
Ackermann
*
and
Lutz
Ackermann
*
Institut für Organsiche und Biomolekulare Chemie and Wöhler Research Institute for Sustainable Chemistry, Georg-August-Universität Göttingen, Tammannstrasse 2, Göttingen 37077, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de; Web: http://www.ackermann.chemie.uni-goettingen.de/ Web: http://wisch.chemie.uni-goettingen.de/
First published on 10th March 2021
Abstract
Herein, we report on selectivity control in C–H activations with alkylidenecyclopropanes (ACPs) for the chemo-selective assembly of cyclopropanes or dienes. Thus, unprecedented rhodaelectro-catalyzed C–H activations were realized with diversely decorated ACPs with a wide substrate scope and electricity as the sole oxidant.
Throughout the last decade, C–H activation has emerged as an increasingly powerful tool in molecular syntheses.1 In sharp contrast, strategies for transition metal-catalyzed C–C activation remain comparably underdeveloped.2 In recent years, major advances, in particular in ring-strain release-promoted C–C cleavages, have been achieved by Dong,3Bower,4 and Marek,5 among others.6 Alkylidenecyclopropanes7 (ACPs) have previously been recognized as a versatile platform for C–H/C–C functionalizations. However, their application within a bifurcated mechanistic manifold for the selective introduction of cyclopropane8 or 1,3-dienes9 motifs has thus far proven elusive, although they represent crucial structural scaffolds in a variety of pharmaceuticals, biologically active molecules and natural products. While a single example of rhodium-catalyzed dienylation was realized with chemical oxidants,10 cyclopropylations are as of yet not available.
The use of electricity to drive chemical reactions has recently witnessed a remarkable renaissance.11 Significant momentum was particularly gained by the merger of metallaelectrocatalysis and C–H activation to avoid often toxic and expensive oxidants.1b,12 With our continued interest in rhodaelectro-catalyzed C–H activation,13 we have now developed a bifurcated C–H activation with alkylidenecyclopropanes that can be conducted under sustainable and operationally-simple electrochemical conditions. Salient features of our strategy include (a) full control of selectivity within a bifurcated manifold for C–H cyclopropylations versus dienylations via β-H over β-C elimination, (b) detailed mechanistic insights by means of experiment and computation, (c) absence of external chemical oxidants, (d) water as the reaction medium, and (e) a user-friendly undivided cell setup without additional electrolyte (Fig. 1).
 | ||
| Fig. 1 Cyclopropylation and dienylation enabled by rhodaelectro-catalysis. | ||
We initiated our studies with indole 1a and ACP 2a to evaluate C–H dienylations and cyclopropylations in a user-friendly undivided cell setup with a graphite felt (GF) anode and a platinum cathode (Table 1). The dienylated product 3aa was obtained in 72% yield in the presence of 2.5 mol% [Cp*RhCl2]2, using 1,4-dioxane/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the solvent. After examination of different bases, NaO2CAd led to the best result, delivering diene 3aa in 85% yield with an Z/E ratio of 4.5/1 (entries 1–5). The indispensable roles of electricity and the rhodium catalyst were further confirmed by control experiments (entries 6 and 7). A variation of the current did not result in an improved performance (entries 8 and 9). We also tested different acids and found that cyclopentanecarboxylic acid proved beneficial (entries 10 and 11). With an increased amount of NaO2CAd, the product was obtained in a higher Z/E ratio, albeit with a small decrease in efficiency (entry 12). A higher reaction temperature improved the efficacy. Importantly, the novel cyclopropylated product 5aa was obtained in high yield when using benzyl ACP 4a.14
:1) as the solvent. After examination of different bases, NaO2CAd led to the best result, delivering diene 3aa in 85% yield with an Z/E ratio of 4.5/1 (entries 1–5). The indispensable roles of electricity and the rhodium catalyst were further confirmed by control experiments (entries 6 and 7). A variation of the current did not result in an improved performance (entries 8 and 9). We also tested different acids and found that cyclopentanecarboxylic acid proved beneficial (entries 10 and 11). With an increased amount of NaO2CAd, the product was obtained in a higher Z/E ratio, albeit with a small decrease in efficiency (entry 12). A higher reaction temperature improved the efficacy. Importantly, the novel cyclopropylated product 5aa was obtained in high yield when using benzyl ACP 4a.14
|
|
||||
|---|---|---|---|---|
| Entry | Base | Acid | Yield (%) | Z/E |
|
a Undivided cell, graphite felt anode (GF), platinum plate cathode (Pt), 1a (0.1 mmol) 2a (0.16 mmol), [Cp*RhCl2]2 (2.5 mol%), base (20 mol%), acid (10 mol%), 1,4-dioxane/H2O (1:1, 4.0 mL), 85 °C, CCE @ 3.0 mA, under air, 4.0 h, yield of isolated product, Z/E ratio determined by 1H NMR spectroscopy, CypCO2H = cyclopentanecarboxylic acid.
b Without electricity, 12 h.
c Without [Cp*RhCl2]2.
d CCE @ 2.0 mA, 6.0 h.
e CCE @ 4.0 mA, 3.0 h.
f NaO2CAd (40 mol%).
g 0.2 mmol scale, 1,4-dioxane/H2O (1:1, 8.0 mL), CCE @ 5.0 mA, 3.0 h.
h 95 °C.
i
4a instead of 2a under the conditions of entry 14.
|
||||
| 1 | NaOAc | CypCO2H | 72 | 3.9/1 |
| 2 | NaOPiv | CypCO2H | 78 | 3.5/1 |
| 3 | NaO2CMes | CypCO2H | 60 | 4.0/1 |
| 4 | NaO2CPh | CypCO2H | 82 | 3.6/1 |
| 5 | NaO2CAd | CypCO2H | 85 | 4.5/1 |
| 6b | NaO2CAd | CypCO2H | 24 | 2.4/1 |
| 7c | NaO2CAd | CypCO2H | — | — |
| 8d | NaO2CAd | CypCO2H | 87 | 3.8/1 |
| 9e | NaO2CAd | CypCO2H | 72 | 3.2/1 |
| 10 | NaO2CAd | MesCO2H | 78 | 3.8/1 |
| 11 | NaO2CAd | PivOH | 82 | 3.3/1 |
| 12f | NaO2CAd | CypCO2H | 82 | 6.0/1 |
| 13fg | NaO2CAd | CypCO2H | 87 | 6.5/1 |
| 14 | NaO 2 CAd | CypCO 2 H | 89 | 7.0/1 |
| 15 | NaO 2 CAd | CypCO 2 H | 95 (5aa) | <1/20 |

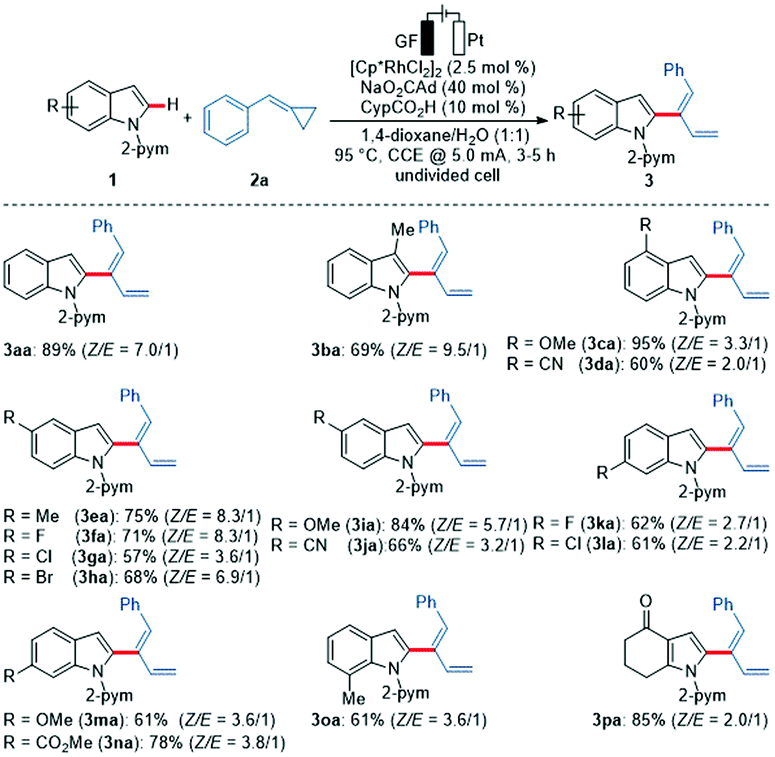
With the optimized reaction conditions for the electrochemical C–H dienylation in hand, its versatility was explored with substituted indoles 1 (Scheme 1). 3-, 5- or 7-Methyl indoles 1 delivered the desired products 3ba, 3ea and 3oa, while the 3-methyl indole 1b gave an improved selectivity. Fluorine- and methoxy-substituted indoles 1 were efficiently transformed, but 6-substituted indoles 1k and 1m displayed a slightly lower efficiency. Various functional groups were tolerated by the rhodium electrocatalyst, such as chloro, bromo and cyano substituents. Interestingly, indole 1n with an ester functionality at the 6-position delivered diene 3na in high yield. The dienylation protocol was also amenable to pyrrole 3pa.15
 | ||
| Scheme 1 Electrocatalytic C–H dienylation of indoles 1. | ||
Next, the robustness of the rhodaelectro-catalyzed C–H dienylation was evaluated with a variety of functionalized cyclopropanes (Scheme 2). Substrates containing bromide groups delivered chemo-selectively the products 3ae and 3am. In contrast to previous studies, electron-deficient heteroarenes showed an inherent high reactivity.13 However, electron-rich substrates also performed well in the electrocatalysis. The connectivity of diene 3ap was unambiguously confirmed by single-crystal X-ray analysis.‡
 | ||
| Scheme 2 Electrochemical C–H dienylation with ACPs 2. | ||
Thereafter, we turned our attention to the versatility of the unprecedented electrochemical C–H cyclopropylation of indoles 1 (Scheme 3). We found that an otherwise reactive hydroxyl was fully tolerated, despite being in close proximity (5ca). Halogen-containing indoles, even the reactive iodo-substituent, were likewise viable substrates. Indoles containing electron-withdrawing or electron-donating groups selectively underwent this transformation. For 7-methyl indole, the cyclopropylation showed a higher efficiency as compared to the dienylation (5oaversus3oa). The rhodaelectrocatalysis proved also applicable to pyrroles, while the structure of the cyclopropylated product 5pa was confirmed by single-crystal X-ray analysis.‡ It is noteworthy that, 2-phenyl pyridine could also be employed for the electrocatalysis to deliver arene 5qa. The tryptamine-derived substrate 1r delivered the challenging ring-opening product 5ra′.
 | ||
| Scheme 3 Electrocatalyzed C–H cyclopropylation of indoles 1, arenes and pyrroles. | ||
Next, we explored the C–H cyclopropylation with differently substituted ACPs 4 (Scheme 4). Substrate 4c bearing an iodo-substituent gave the desired product 5ac with a small amount of the deiodinated product (5aa:5ac 1/3). The aqueous conditions were compatible with linear or branched alkyl-derived cyclopropanes (5ad–5af). The challenging cyclopropane 4g bearing a terminal alkene was also found to be a viable substrate, affording product 5ag in 79% yield. The transformation was also tolerant to changes in the backbone of the cyclic alkanes and generated the desired products 5ah and 5ai. Indeed, the structurally more complex, natural product citronellol-derived starting material 4j was chemo-selectively converted to the desired product 5aj.
 | ||
| Scheme 4 Rhodaelectro-catalyzed C–H cyclopropylation with ACPs 4. | ||
To gain insights into the reaction mechanism, control experiments were performed. The independently prepared cyclometalated complex 916 was found to serve as a catalytically competent species (Scheme 5a). Under the standard conditions but without electricity, H/D exchange of indole 1a with D2O was observed with significant deuterium incorporation at the position C2 (Scheme S2 in the ESI†). However, a significant deuterium-incorporation into product 3aa was not observed, when 1a was reacted with 2a under the electrochemical conditions using D2O as the cosolvent (Scheme S3 in the ESI†). A kinetic isotope effect (KIE) study was next conducted. Parallel independent reactions resulted in a value of kH/kD ≈ 1.4 (Scheme 5b), indicating that the C–H cleavage step is likely not involved in the rate-determining step.14
 | ||
| Scheme 5 Summary of key mechanistic findings. | ||
In order to further understand the catalyst's mode of action, we became interested in studying the rhodaelectro-catalyzed C–H cyclopropylation of indole 1a with ACP 4a by density functional theory (DFT). Geometry optimizations and frequency calculations were performed at the TPSS-D3(BJ)/def2-SVP level of theory, while single point energies were calculated at the PW6B95-D3(BJ)/def2-TZVP+SMD(1,4-dioxane) and PBE0-D3(BJ)/def2-TZVP+SMD(1,4-dioxane) level of theory.14 All energies reported here were calculated at the PW6B95-D3(BJ)/def2-TZVP+SMD(1,4-dioxane)//TPSS-D3(BJ)/def2-SVP level of theory.14 Our calculations indicated that after the migratory insertion of ACP 4a, β-H elimination occurs from the intermediate DviaTS(D-E) (Fig. S1, ESI†) with a barrier of 1.1 kcal mol−1. Moreover, β–H elimination from the intermediate D results in the regioselective formation of the E-isomer as the major product, while the generation of Z-isomer is energetically not favourable.14
Based on our studies, we propose a plausible catalytic cycle for the unprecedented rhodaelectro-C–H-cyclopropylation, which is initiated by the formation of a catalytically competent mononuclear cationic Cp*Rh(III) species. As shown in Fig. 2, coordination of indole 1a to Cp*Rh(III) and facile subsequent cyclorhodation at the 2-position affords rhodacycle A. Then, the insertion of alkene 4a occurs to furnish intermediate D, which undergoes β-H elimination to generate the cyclopropylated product 5aa along with a rhodium(I) intermediate. Finally, the Cp*Rh(III) species is regenerated by rate-limiting reoxidation of rhodium(I) at the anode, while generating molecular hydrogen as the byproduct at the cathode and completing the catalytic cycle. In terms of the dienylation, intermediate D undergoes β-C elimination to form intermediate G (Fig. S10 in the ESI†). Final β-H elimination then delivers the dienylated indole 3aa.
 | ||
| Fig. 2 Proposed mechanism for electro-C–H cyclopropylation with ACPs 4. | ||
In conclusion, we have reported on a versatile rhodaelectro-catalyzed C–H activation with alkylidenecyclopropanes under aqueous conditions, devoid of stoichiometric amounts of chemical oxidants. Our unique strategy allowed for the control of selectivity within a bifurcated mechanistic pathway by the judicious choice of β-H over β-C elimination. Detailed studies by experiment and calculation provided key insights into the catalyst's mode of action, revealing β-H elimination as the key selectivity-determining process for an unprecedented C–H cyclopropylation. The reactive catalyst can be regenerated in a sustainable manner by anodic oxidation, yielding hydrogen as the sole stoichiometric byproduct. Thereby, a wealth of heteroarenes was functionalized with excellent chemo-, position- and diastereoselectivity.
Generous support by the DFG (Gottfried-Wilhelm-Leibniz award to L. A.) and the CSC (fellowship to Z. S.) is gratefully acknowledged. We thank Dr Christopher Golz (Göttingen University) for assistance with the X-ray diffraction analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected recent reviews, see: (a) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed; (b) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed; (c) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (d) C. S. Wang, P. H. Dixneuf and J. F. Soule, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed; (e) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed.
- For selected reviews, see: (a) Y. Cohen, A. Cohen and I. Marek, Chem. Rev., 2021, 121, 140–161 CrossRef CAS PubMed; (b) P.-h. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed; (c) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed; (d) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed.
- (a) Y. Xia, G. Lu, P. Liu and G. Dong, Nature, 2016, 539, 546–550 CrossRef CAS PubMed; (b) T. Xu and G. Dong, Angew. Chem., Int. Ed., 2012, 51, 7567–7571 CrossRef CAS PubMed.
- (a) G.-W. Wang, N. G. McCreanor, M. H. Shaw, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2016, 138, 13501–13504 CrossRef CAS PubMed; (b) M. H. Shaw, E. Y. Melikhova, D. P. Kloer, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2013, 135, 4992–4995 CrossRef CAS PubMed.
- (a) A. Vasseur and I. Marek, Nat. Protoc., 2017, 12, 74–87 CrossRef CAS PubMed; (b) S. R. Roy, D. Didier, A. Kleiner and I. Marek, Chem. Sci., 2016, 7, 5989–5994 RSC; (c) A. Masarwa, D. Didier, T. Zabrodski, M. Schinkel, L. Ackermann and I. Marek, Nature, 2014, 505, 199–203 CrossRef CAS PubMed.
- A review: (a) J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110–139 CrossRef CAS PubMed for selected examples, see: ; (b) H. Wang, I. Choi, T. Rogge, N. Kaplaneris and L. Ackermann, Nat. Catal., 2018, 1, 993–1001 CrossRef CAS; (c) S. Okumura, F. Sun, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2017, 139, 12414–12417 CrossRef CAS PubMed; (d) E. Ozkal, B. Cacherat and B. Morandi, ACS Catal., 2015, 5, 6458–6462 CrossRef CAS; (e) H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2011, 133, 15244–15247 CrossRef CAS PubMed; (f) L. J. Gooßen, G. Deng and L. M. Levy, Science, 2006, 313, 662–664 CrossRef PubMed.
- For selected reviews, see: (a) L. Yu, M. Liu, F. Chen and Q. Xu, Org. Biomol. Chem., 2015, 13, 8379–8392 RSC; (b) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913–924 CrossRef CAS PubMed; (c) A. de Meijere, S. I. Kozhushkov and H. Schill, Chem. Rev., 2006, 106, 4926–4996 CrossRef CAS PubMed.
- (a) L. A. Maslovskaya, A. I. Savchenko, C. J. Pierce, G. M. Boyle, V. A. Gordon, P. W. Reddell, P. G. Parsons and C. M. Williams, Chem. – Eur. J., 2019, 25, 1525–1534 CrossRef CAS PubMed; (b) D. Y. K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631–4642 RSC.
- (a) Y.-Q. Zhu, Y.-X. Niu, L.-W. Hui, J.-L. He and K. Zhu, Adv. Synth. Catal., 2019, 361, 2897–2903 CrossRef CAS; (b) R. Feng, J. A. Smith and K. D. Moeller, Acc. Chem. Res., 2017, 50, 2346–2352 CrossRef CAS PubMed; (c) A. Misale, S. Niyomchon and N. Maulide, Acc. Chem. Res., 2016, 49, 2444–2458 CrossRef CAS PubMed; (d) S. Carosso and M. J. Miller, Org. Biomol. Chem., 2014, 12, 7445–7468 RSC; (e) W. Erb and J. Zhu, Nat. Prod. Rep., 2013, 30, 161–174 RSC.
- R. Liu, Y. Wei and M. Shi, Chem. Commun., 2019, 55, 7558–7561 RSC.
- (a) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chemistry, 2020, 6, 2484–2496 CrossRef CAS; (b) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC; (c) M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC; (d) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (e) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (f) T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Trends Chem., 2019, 1, 63–76 CrossRef CAS; (g) R. D. Little and K. D. Moeller, Chem. Rev., 2018, 118, 4483–4484 CrossRef CAS PubMed; (h) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed; (i) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed; (j) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (k) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- (a) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed; (b) P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, Chem. Soc. Rev., 2020, 49, 4254–4272 RSC.
- (a) Y. Zhang, J. Struwe and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 15076–15080 CrossRef CAS PubMed; (b) W.-J. Kong, Z. Shen, L. H. Finger and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 5551–5556 CrossRef CAS PubMed; (c) Z.-J. Wu, F. Su, W. Lin, J. Song, T.-B. Wen, H.-J. Zhang and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 16770–16774 CrossRef CAS PubMed; (d) W.-J. Kong, L. H. Finger, A. M. Messinis, R. Kuniyil, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 17198–17206 CrossRef CAS PubMed.
- Detailed information is given in the ESI†.
- Under otherwise identical reaction conditions, alternative heterocycles thus far led to unsatisfactory results.
- X. Zhou, Y. Pan and X. Li, Angew. Chem., Int. Ed., 2017, 56, 8163–8167 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2025011 (3ap) and 2025012 (5pa). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc08123j |
| ‡ Deposition numbers 2025011 (3ap) and 2025012 (5pa) contain the supplementary crystallographic data for this paper. |
| This journal is © The Royal Society of Chemistry 2021 |