
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChorismate- and isochorismate converting enzymes: versatile catalysts acting on an important metabolic node†
Florian
Hubrich
 *a,
Michael
Müller
b and
Jennifer N.
Andexer
*b
*a,
Michael
Müller
b and
Jennifer N.
Andexer
*b
aETH Zurich, Institute of Microbiology, Vladimir-Prelog-Weg 4, 8093 Zurich, Switzerland. E-mail: fhubrich@biol.ethz.ch
bAlbert-Ludwigs-University Freiburg, Institute of Pharmaceutical Sciences, Albertstrasse 25, 79104 Freiburg, Germany. E-mail: jennifer.andexer@pharmazie.uni-freiburg.de
First published on 19th February 2021
Abstract
Chorismate and isochorismate represent an important branching point connecting primary and secondary metabolism in bacteria, fungi, archaea and plants. Chorismate- and isochorismate-converting enzymes are potential targets for new bioactive compounds, as well as valuable biocatalysts for the in vivo and in vitro synthesis of fine chemicals. The diversity of the products of chorismate- and isochorismate-converting enzymes is reflected in the enzymatic three-dimensional structures and molecular mechanisms. Due to the high reactivity of chorismate and its derivatives, these enzymes have evolved to be accurately tailored to their respective reaction; at the same time, many of them exhibit a fascinating flexibility regarding side reactions and acceptance of alternative substrates. Here, we give an overview of the different (sub)families of chorismate- and isochorismate-converting enzymes, their molecular mechanisms, and three-dimensional structures. In addition, we highlight important results of mutagenetic approaches that generate a broader understanding of the influence of distinct active site residues for product formation and the conversion of one subfamily into another. Based on this, we discuss to what extent the recent advances in the field might influence the general mechanistic understanding of chorismate- and isochorismate-converting enzymes. Recent discoveries of new chorismate-derived products and pathways, as well as biocatalytic conversions of non-physiological substrates, highlight how this vast field is expected to continue developing in the future.
 Florian Hubrich | Florian Hubrich studied chemistry and biology at the University of Freiburg. Following his Staatsexamen he started his PhD in chemical biology with focus on chorismatases supervised by Jennifer Andexer and Michael Müller at the Institute of Pharmaceutical Sciences in Freiburg and finished in 2014. In 2015 he moved to Switzerland where he worked for the BACHEM AG as project chemist and group leader for UHPLC analysis of peptides. In 2018 he returned to academia and joined Jörn Piel's lab as a postdoctoral researcher at the ETH Zurich. Currently, he studies the biosynthesis of peptide natural products from ribosomal origin. |
 Michael Müller | Michael Müller studied chemistry at the University of Bonn. After receiving the diploma degree, he began his PhD at the Ludwig-Maximilians-Universität in Munich under the guidance of Professor Steglich, finishing in 1995. Following a research exchange at the University of Washington working, he became the Group Leader of Bioorganic Chemistry at the Forschungszentrum Jülich. Prof. Müller qualified as a University Lecturer for Organic and Bioorganic Chemistry in 2002 and was appointed the Professorship for Pharmaceutical and Medicinal Chemistry at the Albert-Ludwigs-Universtät Freiburg in 2004. His research interests include chemoenzymatic synthesis, natural products, asymmetric and diversity-oriented synthesis as well as sustainability. |
 Jennifer N. Andexer | Following a diploma in Biology, Jennifer Andexer carried out her doctoral research working on oxynitrilases at the Institute of Molecular Enzyme Technology (University of Duesseldorf) supervised by Thorsten Eggert, Karl-Erich Jaeger and Martina Pohl. In 2008, she moved to the labs of Joe Spencer, Finian Leeper and Peter Leadlay (University of Cambridge), studying biosynthetic pathways of natural products. She has been head of the Chemical Biology group (Institute of Pharmaceutical Sciences, University of Freiburg) since 2011 and was appointed Heisenberg Professor of Pharmaceutical and Medicinal Chemistry in 2020. She works on enzyme selectivity, cofactor regeneration systems and cofactor analogues. |
1. Introduction
In 2001, Dosselaere and Vanderleyden described chorismate (1) as a “metabolic node in action” in an outstanding review of the five most important families of chorismate-converting enzymes in microorganisms.1 At that time, eight products of immediate chorismate (1) and isochorismate (2) origin‡ were known, among them the amino derivatives 2-amino-2-deoxyisochorismate (3, ADIC) and 4-amino-4-deoxychorismate (4, ADC), which may be considered equally versatile branching points, as discussed below (Section 13).1–5 Within the last two decades, this number of products from a single metabolic substrate has increased to at least twelve (amino/iso)chorismate derivatives. In addition, the knowledge of the structural basis, the molecular mechanisms of the enzymes involved, and the regulation of product formation has grown enormously.Here, we present an update on the structural basis and underlying molecular mechanisms of the entire class of chorismate- and isochorismate-converting enzymes.§ Rather than providing an exhaustive compilation of original experiments, we aimed to prepare a general overview serving as a starting point for research into the field and as a complement to a wealth of prior subfamily- and subtopic-specific reviews.6–13
In addition, recent advances in the field and their potential influence on the understanding of chorismate-converting enzymes in general are highlighted, followed by a brief overview of novel chorismate-derived natural products and biocatalytic applications of chorismate-converting enzymes.
1.1. Chorismate and isochorismate: highly reactive molecules with an enormous potential for diversity-oriented product formation
Chorismate (1) and its isomer isochorismate (2, 2-hydroxy-4-deoxychorismate) feature several chemical moieties attached to its central cyclohexadiene system: a hydroxyl and a carboxyl group as well as an enolpyruvyl ether. These moieties make (iso)chorismate highly reactive under standard conditions and challenging to handle. Notably, the reactivity of (iso)chorismate is a prerequisite for ‘diversity-oriented’ product formation in enzymatic and non-enzymatic reactions, as studied intensively since the 1960s.14–20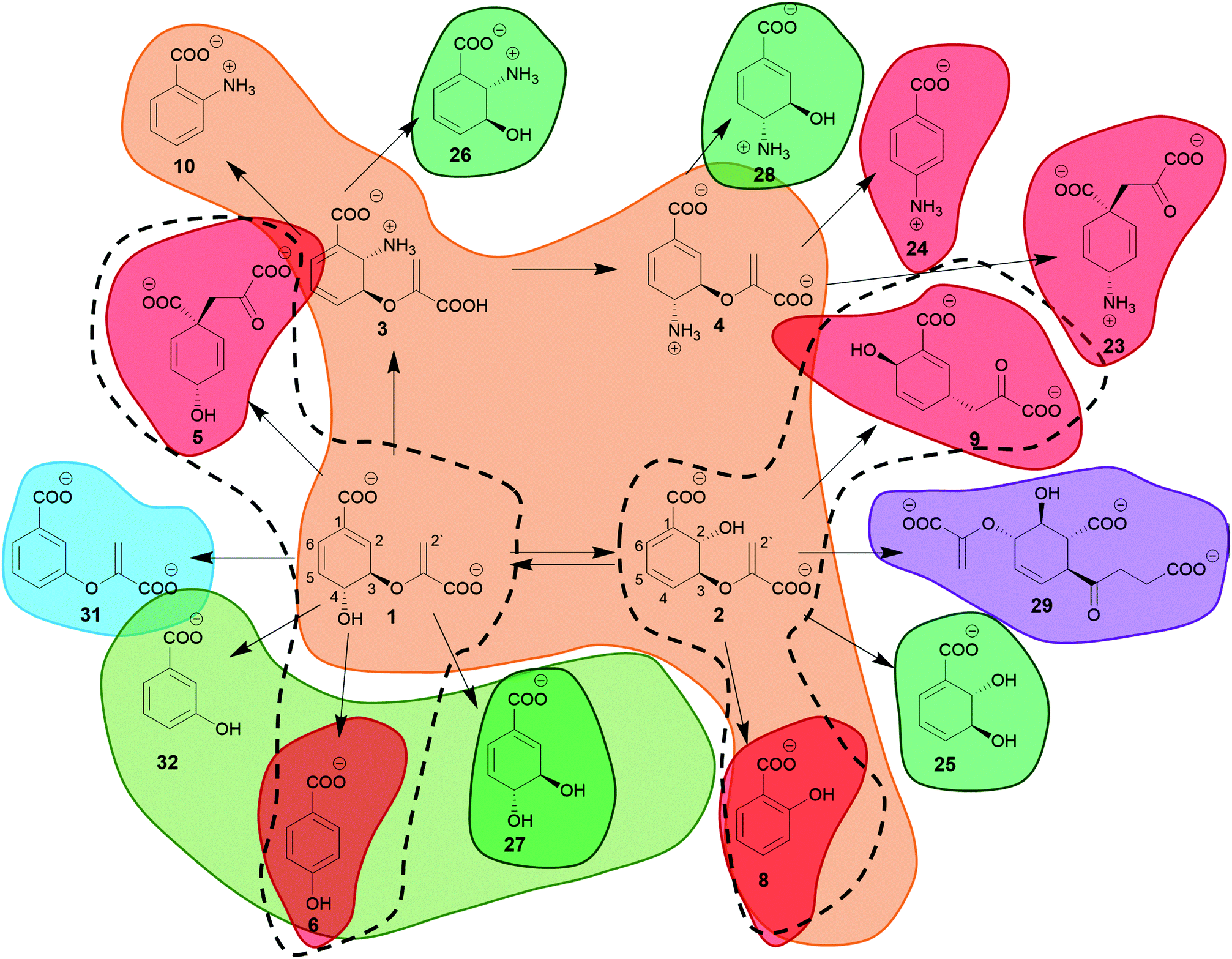 | ||
| Fig. 1 Chorismate (1), isochorismate (2), and (iso)chorismate derivatives. The colours indicate the different families of chorismate-converting enzymes that catalyse the respective reaction: orange – MST (involved in the biosynthesis of menaquinone, siderophores and tryptophan) enzymes, red – SPR (catalysing solely pericyclic reactions) enzymes, green – isochorismatases, olive – chorismatases, blue – chorismate dehydratases, purple – SEPHCHC synthases, dashed lines – non-enzymatic reactions. | ||
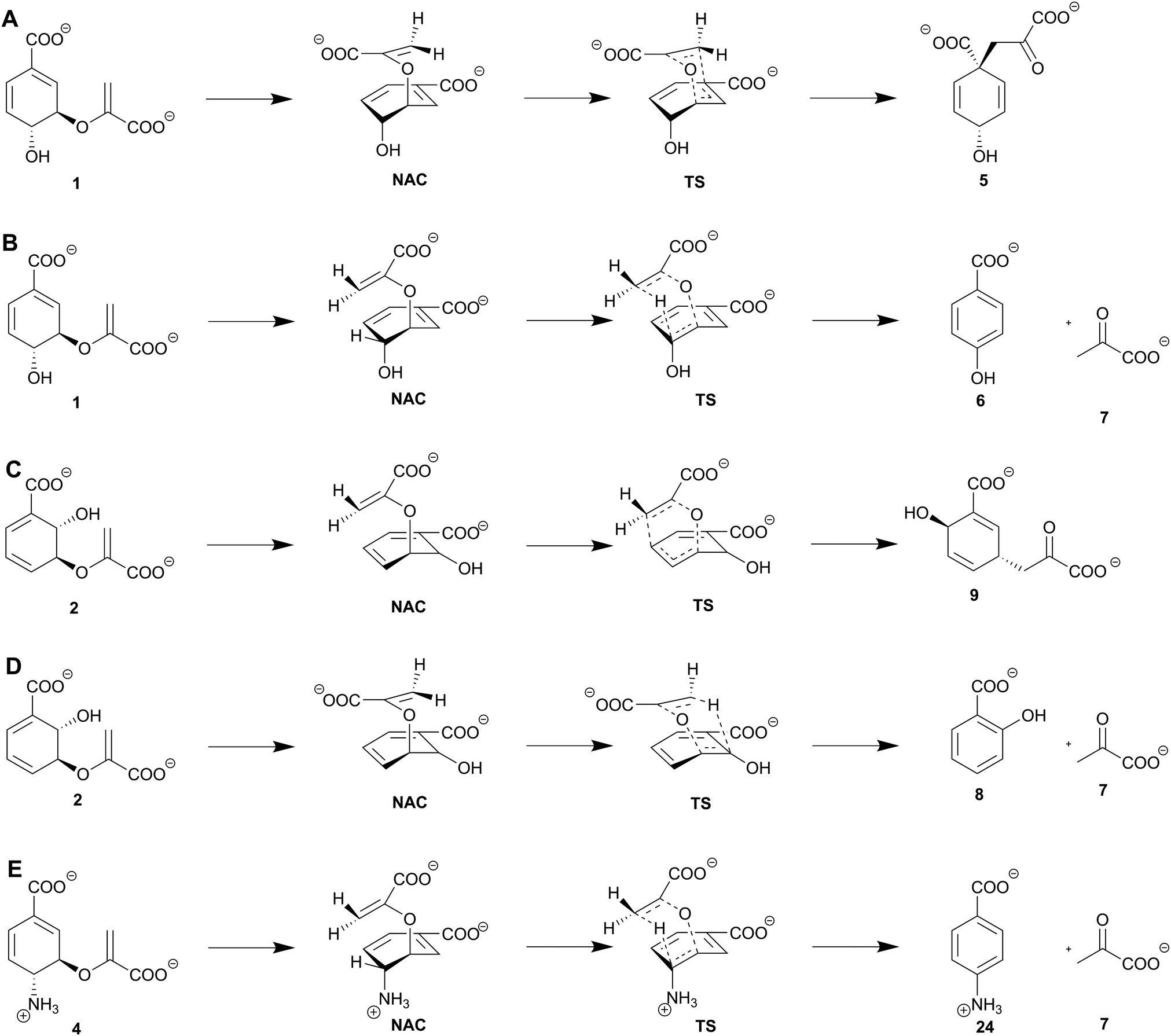 | ||
| Scheme 1 Non-enzymatic reactions of chorismate and isochorismate, and molecular mechanisms of SPR enzymes, including near attack conformation (NAC) and transition state (TS). (A) [3,3]-Sigmatropic rearrangement of chorismate (1) to prephenate (5) (non-enzymatic and by chorismate mutases). (B) [1,5]-Sigmatropic rearrangement of chorismate (1) to 4-HBA (6) and pyruvate (7) (non-enzymatic and by chorismate lyases). (C) [3,3]-Sigmatropic rearrangement of isochorismate (2) to isoprephenate (9) (non-enzymatic). (D) [1,5]-Sigmatropic rearrangement of isochorismate (2) to salicylate (8) and pyruvate (7) (non-enzymatic and by isochorismate-pyruvate lyases). (E) Non-natural [1,5]-sigmatropic rearrangement of 4-amino-4-deoxychorismate (4) to pABA (24) and pyruvate (7) (by chorismate lyases). | ||
Data from experiments addressing the kinetic isotope effect via nuclear magnetic resonance (NMR) spectroscopy in combination with computational calculations indicate that both reactions proceed via a pseudo-diaxial chair-like transition state (TS), although the concerted mechanism is highly asynchronous (Scheme 1).18,19,21 Quantum mechanics and molecular mechanics calculations (QM/MM) support the pericyclic nature of the reactions in solution. Nevertheless, the rate-limiting step of the reactions, including their enzymatic counterparts, is still under discussion.22–26 Hilvert and co-workers determined that the thermal stability of chorismate at 60 °C is higher than the thermal stability of isochorismate.18,19,21 Comparable findings were obtained by us with chorismate and isochorismate at 25 °C; however, this study further suggested that the half-life time strongly depends on the purity of the compounds.27 The molar enthalpy of the reaction from chorismate to isochorismate in solution was determined as 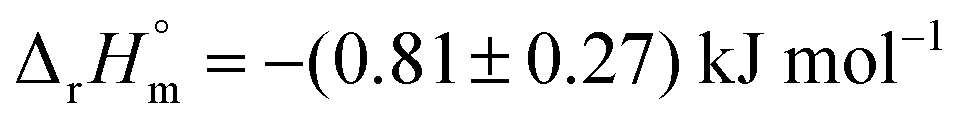 by the Goldberg group.28
by the Goldberg group.28
1.1.3 Chorismate as a central branching point in metabolism. In vivo, chorismate is the formal endpoint of the shikimate biosynthetic pathway and a central branching point connecting the primary and the secondary metabolism in bacteria, plants, and fungi.1,34,35
Starting from chorismate or isochorismate, important primary and secondary metabolites such as ubiquinone (11), menaquinone (12), siderophores [e.g., enterobactin (13)], folates (14), and the aromatic amino acids tryptophan (Trp, 15), phenylalanine (Phe, 16), and tyrosine (Tyr, 17) are synthesised (Fig. 2).3,36 A wide range of bioactive compounds is accessible via biosynthetic pathways branching from (iso)chorismate or using (iso)chorismate derivatives as starter units for thiotemplate biosynthesis, e.g. of stravidin (18, antibiotic), rapamycin (19, immunosuppressant), and unantimycins (20, anticancer agents), or for terpenoids such as merosterols (21, cytotoxic) (Fig. 2).37–40 As the shikimate and the corresponding branching biosynthetic pathways are not present in the Animalia kingdom including humans, these pathways and the enzymes involved are promising targets for the development of selective drugs and pesticides.1,34 Nevertheless, there are hints that shikimate-like pathways might be present in this phylum as summarised in a recent review on the natural product potential of the animal world.41
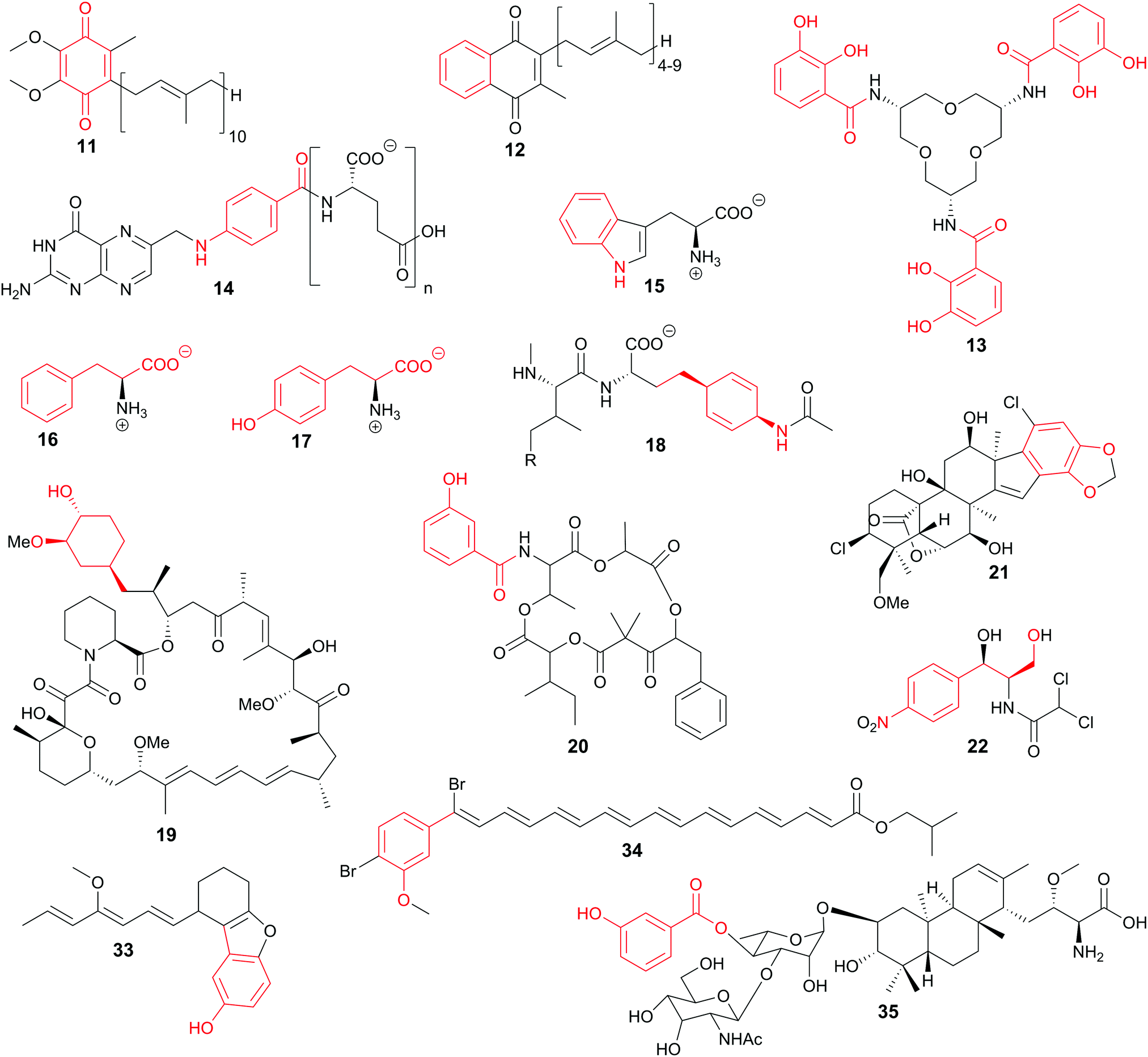 | ||
| Fig. 2 Selected natural products and primary metabolites of (iso)chorismate origin: ubiquinone (11), menaquinone (12), enterobactin (13), folate (14), tryptophan (15), phenylalanine (16), tyrosine (17), stravidin (18), rapamycin (19), unantimycin A (20), merosterol A (21), chloramphenicol (22), cuevaene A (33), xanthomonadin I (34), brasilicardin (35). (Iso-)chorismate-derived moieties are red. | ||
2. A handful of enzymes access a plethora of products by a vast number of molecular mechanisms
Due to its role as a metabolic branching point, (iso)chorismate serves as a substrate for many different enzymes. Therefore, it is not surprising that non-enzymatic reactions and products of (iso)chorismate in solution have enzymatic counterparts. Fig. 2 shows a selection of products formed from (iso)chorismate. Remarkably, structural studies on chorismate-converting enzymes revealed that only a few different enzyme folds provide access to this vast range of products (Fig. 3 and 4). In some cases, one subfamily with the same enzyme fold catalyses the formation of different products, while other examples are known where two subfamilies featuring different enzyme folds provide the same product (Fig. 1 and Table 1). The structural diversity of chorismate-converting enzymes (Fig. 3 and 4) is echoed in the diversity of catalytic mechanisms (Schemes 2–7).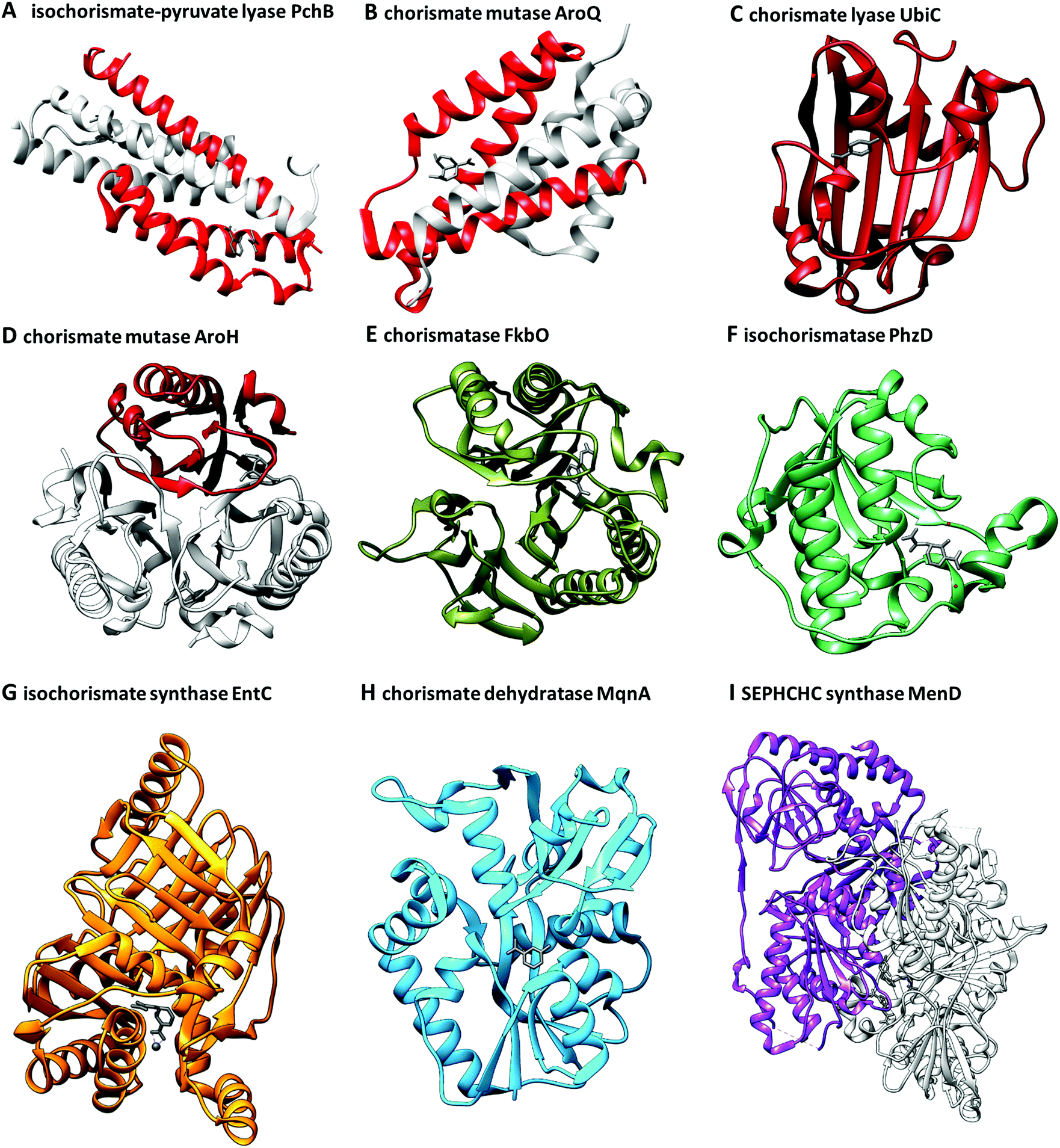 | ||
| Fig. 3 Three-dimensional folds of chorismate-converting enzymes in complex with substrates, products, transition state analogues, or competitive inhibitors (grey). All structures are presented in their catalytic essential forms; for oligomers a single subunit is coloured analogously to Fig. 1: orange – MST enzymes, red – SPR enzymes, green – isochorismatases, olive – chorismatases, blue – chorismate dehydratases, purple – SEPHCHC synthases. (A) Homodimeric isochorismate-pyruvate lyase PchB with co-crystallised salicylate (8) and pyruvate (7) (PDB: 3REM). (B) Homodimeric chorismate mutase AroQ with co-crystallised 8-hydroxy-2-oxa-bicyclo[3.3.1]non-6-ene-3,5-dicarboxylate (PDB: 2FP2). (C) Monomeric chorismate lyase UbiC with co-crystallised 4-HBA (6) (PDB: 1TT8). (D) Monotrimeric chorismate mutase AroH with co-crystallised 8-hydroxy-2-oxa-bicyclo[3.3.1]non-6-ene-3,5-dicarboxylate (PDB: 3ZP4). (E) Monomeric chorismatase FkbO with co-crystallised 3-(2-carboxyethyl)benzoate (PDB: 4BPS). (F) Monomer of the homodimeric isochorismatase PhzD with co-crystallised ADIC (3) (PDB: 3R77). (G) Monomeric isochorismate synthase EntC with co-crystallised isochorismate (2) and magnesium ion (PDB: 3HWO). (H) Monomer of the homodimeric chorismate dehydratase MqnA with co-crystallised 3-HBA (32) (PDB: 6O9A). (I) Dimer of the homotetrameric SEPHCHC synthase MenD with co-crystallised magnesium ion, thiamine diphosphate and isochorismate (2) (PDB: 5ESO). | ||
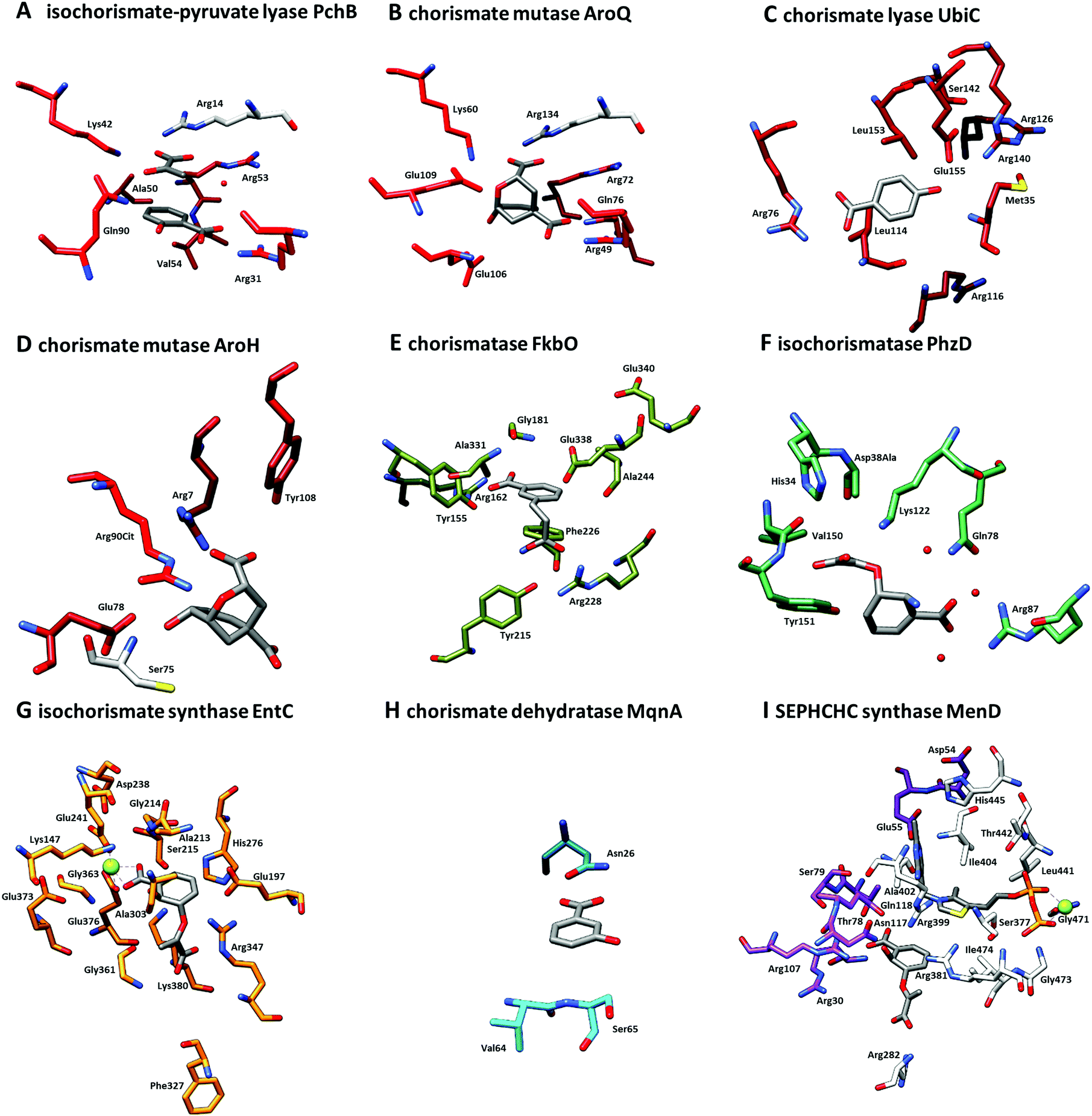 | ||
| Fig. 4 Close-up of active sites from structures shown in Fig. 3. (A) Isochorismate-pyruvate lyase PchB with co-crystallised salicylate (8) and pyruvate (7) (PDB: 3REM). (B) Chorismate mutase AroQ with co-crystallised 8-hydroxy-2-oxa-bicyclo[3.3.1]non-6-ene-3,5-dicarboxylate (PDB: 2FP2). (C) Chorismate lyase UbiC with co-crystallised 4-HBA (6) (PDB: 1TT8). (D) Chorismate mutase AroH with co-crystallised 8-hydroxy-2-oxa-bicyclo[3.3.1]non-6-ene-3,5-dicarboxylate (PDB: 3ZP4). (E) Chorismatase FkbO with co-crystallised 3-(2-carboxyethyl)benzoate (PDB: 4BPS). (F) Isochorismatase PhzD with co-crystallised ADIC (3) (PDB: 3R77). (G) Isochorismate synthase EntC with co-crystallised isochorismate (2) and magnesium ion (PDB: 3HWO). (H) Chorismate dehydratase MqnA with co-crystallised 3-HBA (32) (PDB: 6O9A). (I) SEPHCHC synthase MenD with co-crystallised magnesium ion, thiamine diphosphate and isochorismate (2) (PDB: 5ESO). Amino acid residues located on different quarternary structure elements are shown in colour and grey. | ||
| Classification | Example | EC number | Enzyme fold | Molecular mechanism(s) | Product(s) | ||
|---|---|---|---|---|---|---|---|
| Family | Subfamily | Name (accession number) | Organism | ||||
| SPR enzymes | AroH-chorismate mutases | AroH (P19080) | Bacillus subtilis | 5.4.99.5 | AroH-fold | [3,3]-Sigmatropic rearrangement | Prephenate (5) |
| AroQ-chorismate mutases | AroQ (Q9HU05) | Escherichia coli | 5.4.99.5 | AroQ-fold | [3,3]-Sigmatropic rearrangement | Prephenate (5) | |
| Isochorismate-pyruvate lyases | PchB (Q51507) | Pseudomonas aeruginosa | 4.2.99.21 | [1,5]-Sigmatropic rearrangement | Salicylate (8) + pyruvate (7) | ||
| Chorismate lyases | UbiC (P26602) | Escherichia coli | 4.1.3.40 | UbiC-fold | [1,5]-Sigmatropic rearrangement | 4-HBA (6) + pyruvate (7) | |
| Chorismate-pyruvate lyases | |||||||
| MST enzymes | Isochorismate synthases | EntC (P0AEJ2) | Escherichia coli | 5.4.4.2 | MST-fold | Nucleophilic substitution (SN2) | Isochorismate (2) |
| Salicylate synthases | MbtI (P9WFX1) | Mycobacterium tuberculosis | 5.4.4.2 | Nucleophilic substitution (SN2) | Salicylate (8) + pyruvate (7) | ||
| 4.2.99.21 | [1,5]-Sigmatropic rearrangement | ||||||
| 2-Amino-2-deoxyisochorismate (ADIC) synthases | PhzE (G3XCK6) | Pseudomonas aeruginosa | 2.6.1.86 | Nucleophilic substitution (SN2) | 2-Amino-2-deoxyisochorismate (ADIC) (3) | ||
| Anthranilate synthase | TrpE (P00897) | Serratia marcescens | 4.1.3.27 | Nucleophilic substitution (SN2) | Anthranilate (10) + pyruvate (7) | ||
| [1,5]-Sigmatropic rearrangement | |||||||
| 4-Amino-4-deoxychorismate (ADC) synthases | PabA (P06193) | Stenotrophomonas maltophilia | 2.6.1.85 | 2× nucleophilic substitution (SN2) | 4-Amino-4-deoxychorismate (ADC) (4) | ||
| Isochorismatases | Isochorismatases | EntB (P0ADI4) | Escherichia coli | 3.3.2.1 | IHL-fold | Vinyl ether hydrolysis | 2,3-trans-CHD (25)/2,3-trans-CHA (26)/3,4-trans-CHD (25)/3,4-trans-CHA (28) + pyruvate (7) |
| SEPHCHC synthases | SEPHCHC synthases | MenD (P17109) | Escherichia coli | 2.2.1.9 | ThDP-fold | 1,4-Conjugate addition | SEPHCHC (29) |
| Chorismate dehydratases | Chorismate dehydratases | MqnA (Q5SK49) | Deinococcus radiodurans | 4.2.1.151 | MqnA-fold | Elimination (E1cb) | Enoyl benzoate (31) |
| Chorismatases | Type-I chorismatases | FkbO (Q9KID9) | Streptomyces hygroscopicus subsp. ascomyceticus | 3.3.2.13 | Toblerone-fold | Vinyl ether hydrolysis | 3,4-trans-CHD (27) + pyruvate (7) |
| Type-II chorismatases | Hyg5 (O30478) | Streptomyces hygroscopicus subsp. hygroscopicus | 4.1.3.45 | Nucleophilic substitution (arene oxide intermediate) | 3-HBA (32) + pyruvate (7) | ||
| Type-III chorismatases | XanB2 (Q3I352) | Xanthomonas campestris pv. campestris | 4.1.3.40/45 | Nucleophilic substitution (arene oxide intermediate) | 3-HBA (32)/4-HBA (6) + pyruvate (7) | ||
| Type-IV chorismatases | SmCH-IV (WP_051010542.1/NCBI ID) | Stenotrophomonas maltophilia | 4.1.3.40 | Nucleophilic substitution (arene oxide intermediate) | 4-HBA (6) + pyruvate (7) | ||
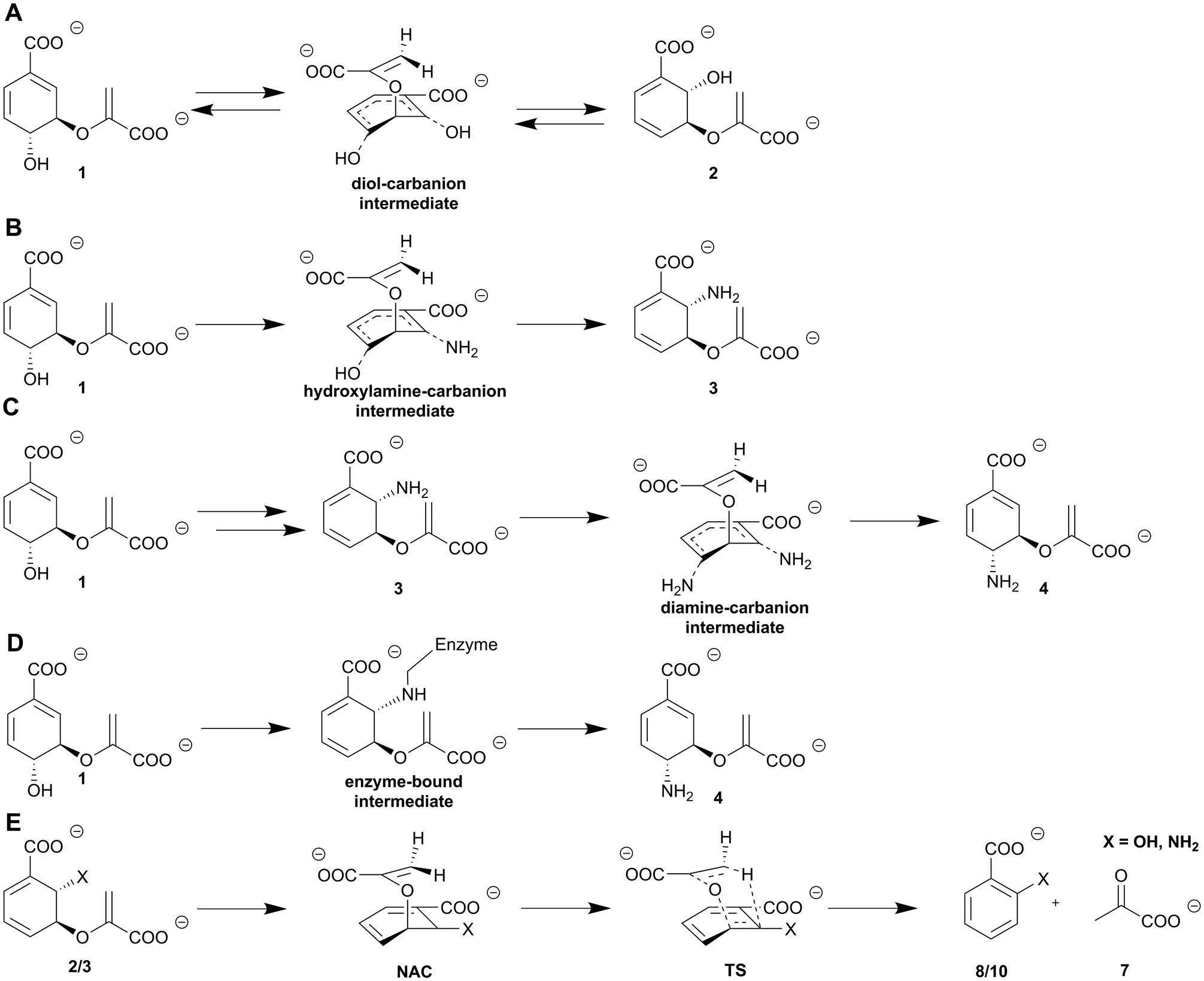 | ||
| Scheme 2 Molecular mechanisms of MST enzymes. (A) SN2-type mechanism including diol-carbanion intermediate from chorismate (1) to isochorismate (2) catalysed by isochorismate synthases. (B) SN2-type mechanism including hydroxylamine-carbanion intermediate from chorismate (1) to ADIC (3) catalysed by ADIC synthases. (C) SN2-type mechanism including diamine-carbanion intermediate from chorismate (1) via ADIC as an intermediate (3) to ADC (4) catalysed by ADC synthases. (D) SN2-type mechanism including enzyme-bound intermediate from chorismate (1) to ADC (4) of ADC synthases. (E) Lyase reaction of isochorismate (2, X = OH) or ADIC (3, X = NH2) to salicylate (8, X = OH) or anthranilate (10, X = NH2), respectively, and pyruvate (7) including near attack conformation (NAC) and transition state (TS). | ||
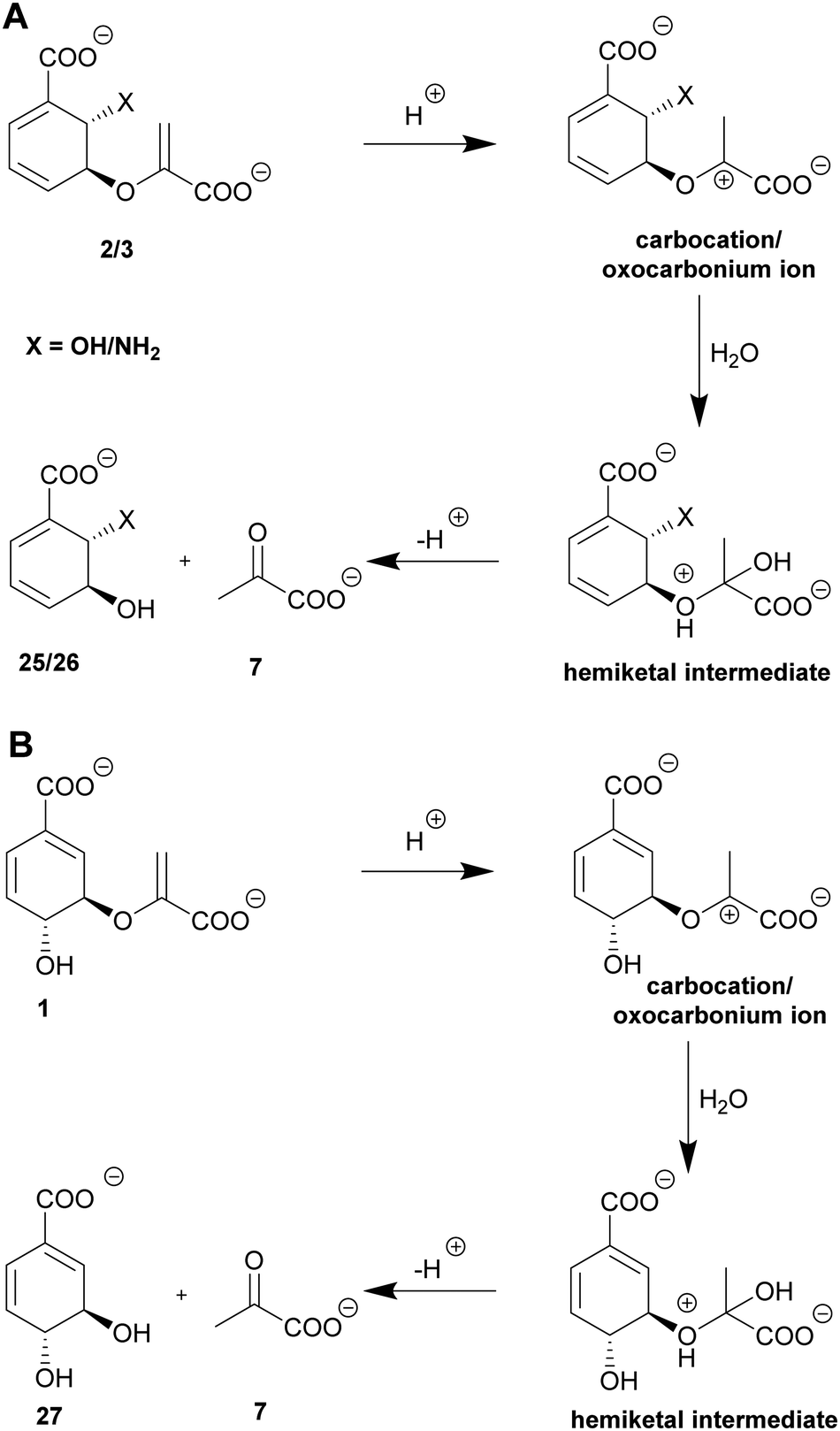 | ||
| Scheme 3 Suggested molecular mechanisms of isochorismatases and type-I chorismatases. (A) Vinyl ether hydrolysis of isochorismate (2) and ADIC (3) to 2,3-trans-CHD (25) and 2,3-trans-CHA (26), respectively, and pyruvate (7) via a carbocation/oxocarbonium ion and an unstable hemiketal intermediate of isochorismatases. (B) Vinyl ether hydrolysis of chorismate (1) to 3,4-trans-CHD (27) and pyruvate (7) via the same mechanism proposed for chorismatases. | ||
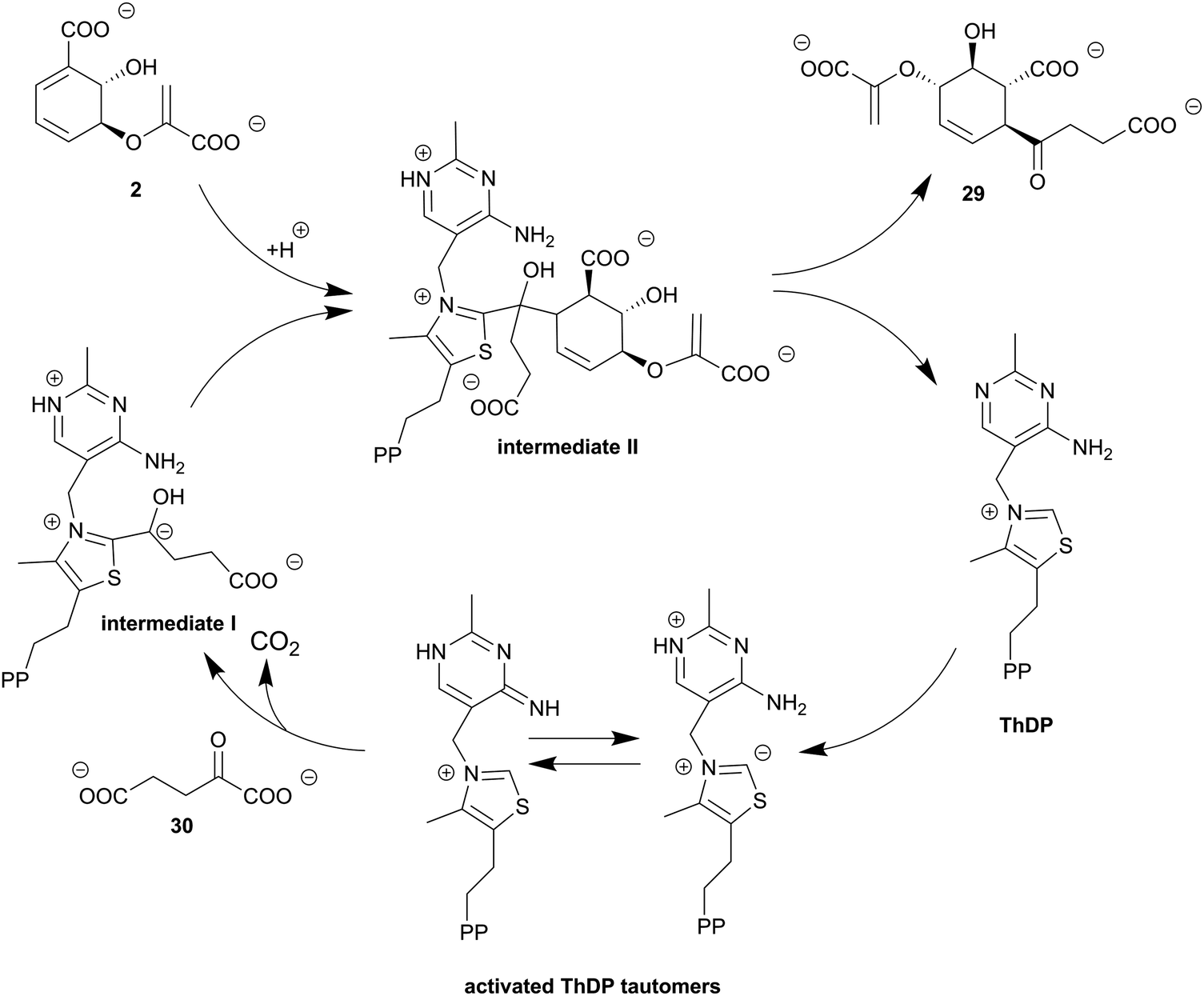 | ||
| Scheme 4 1,4-Conjugate addition of SEPHCHC synthases. ThDP forms intermediate I after addition at C2 of 2-oxoglutarate (30) and decarboxylation of the C1 carboxyl group. Subsequent addition of isochorismate (2) leads to intermediate II before SEPHCHC (29) is released with regeneration of ThDP. Figure adapted from ref. 105 | ||
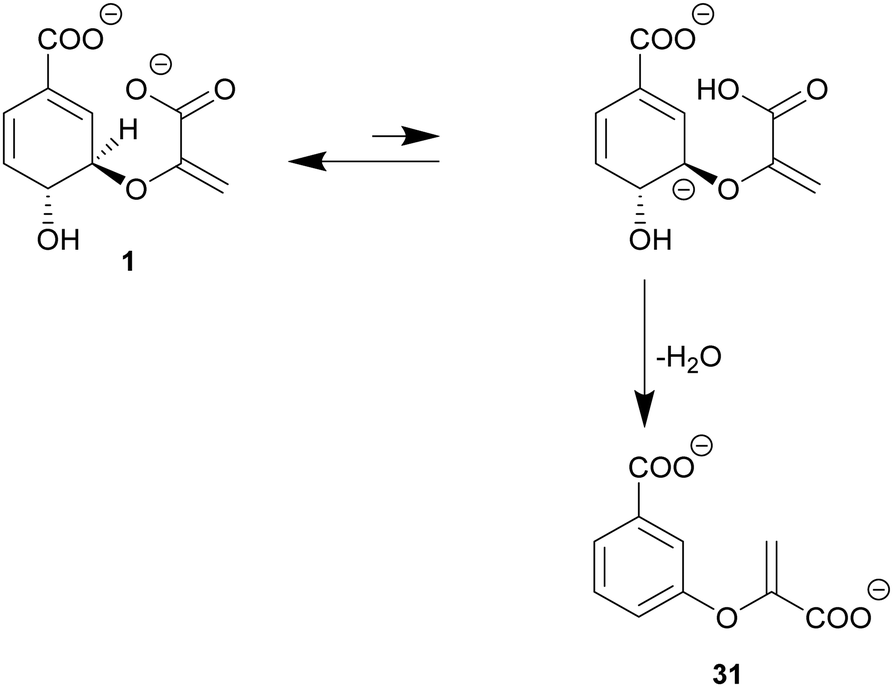 | ||
| Scheme 5 Modified E1cb mechanism of chorismate dehydratases. The carboxylate of the enolpyruvyl tail deprotonates chorismate (1) at C3 of the cyclohexadiene ring. Subsequently, the C4-hydroxyl group serves as a leaving group, with aromatisation of the cyclohexadiene ring leading to 3-enoylpyruvyl benzoate (31). | ||
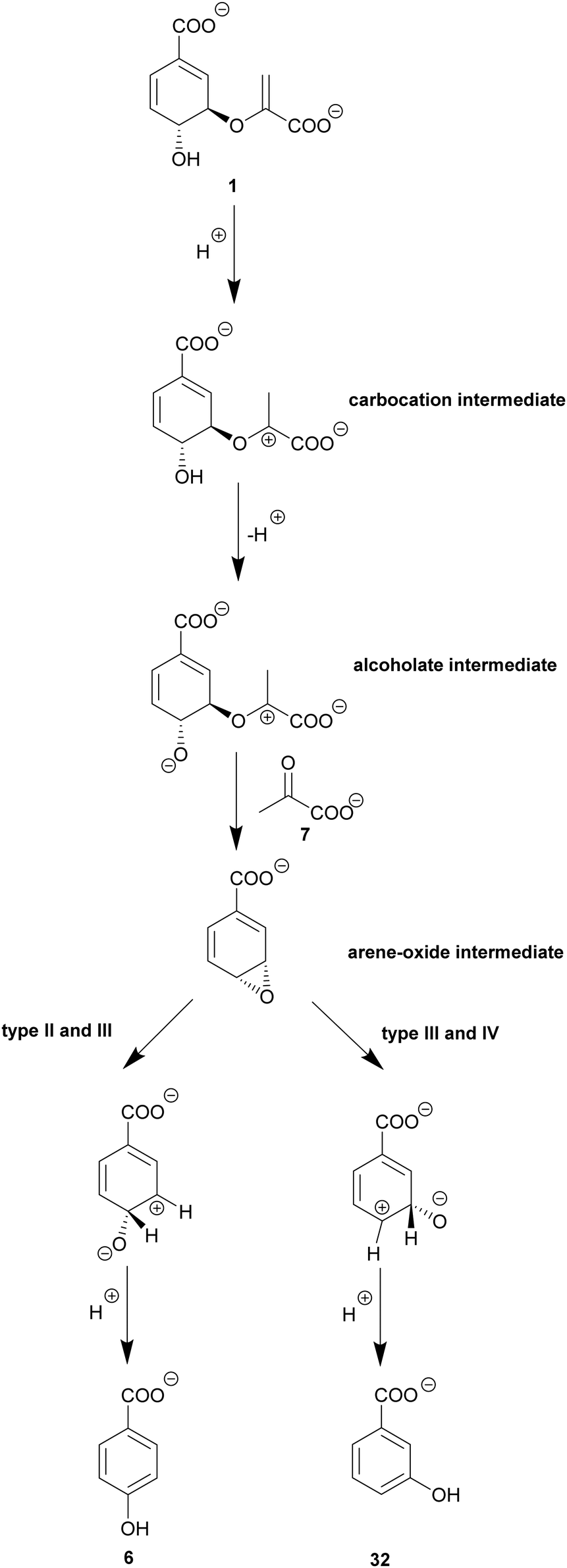 | ||
| Scheme 6 Putative molecular mechanisms of type-II, -III, and -IV chorismatases. Starting from chorismate (1), a carbocation/oxocarbonium ion is formed, followed by formation of an alcoholate at C4 and subsequent attack at C3 leading to an arene oxide intermediate with concomitant loss of pyruvate (7). The ring opening via an NIH shift determines the products 3-HBA (32) and/or 4-HBA (6) depending on the chorismatase subfamily. | ||
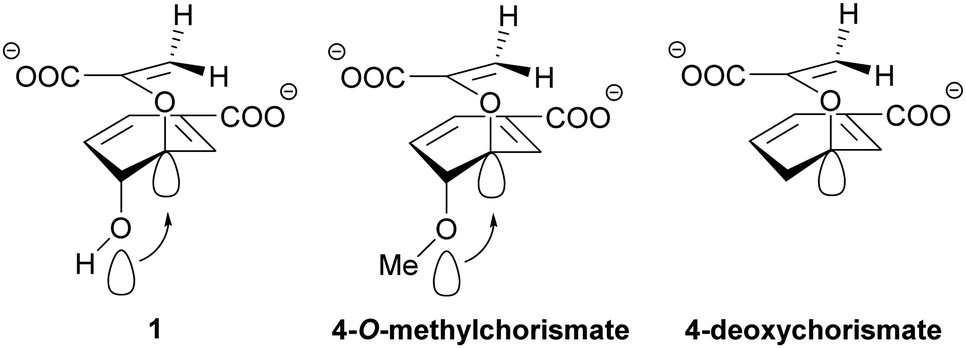 | ||
| Scheme 7 Anomeric effect in chorismate derivatives. The free orbitals of the C4–oxygen of chorismate (1) cause an anomeric effect that weakens the C–O bond at C3 promoting bond breakage and product formation. An additional methyl group at the C4–oxygen increases this effect in 4-O-methylchorismate while the effect is absent in 4-deoxychorismate. This highlights the importance of the C4-hydroxyl group for product formation in chorismate-converting enzymes in general. | ||
The majority of molecular mechanisms in chorismate-converting enzymes are either pericyclic reactions or reactions requiring acid–base catalysis. Nonetheless, several chorismate-converting enzymes are suggested to catalyse both reaction types in the same active site (Table 1). Pericyclic reactions are fascinating reactions in general; nevertheless, seemingly common acid–base reactions catalysed by chorismate-converting enzymes often follow sophisticated routes. One major factor determining product formation in chorismate-converting enzymes are electrostatic effects caused by amino acid residues of the active site and the so-called secondary shell.42 These electrostatic effects contribute at various time points to the reaction trajectory. They usually tightly lock the substrate in a pseudo-diaxial conformation that promotes proper product formation by stabilising distinct transition states or intermediates, and thus determine product specificity and selectivity. Based on the structural knowledge of chorismate-converting enzymes, a broader understanding of product formation has been one main focus of studies for the last two decades.7,21,29,43–47 Approaches using labelled substrates and/or solvents, rational site-directed mutagenesis, and inhibition studies are just a few examples of how open questions were resolved.7,18,19,21,29,47–49 The ‘holy grail’ of these studies is often considered the conversion of the activity of a chorismate-converting enzyme of one (sub)family into one of another subfamily. So far, most of these attempts were unsuccessful, or accompanied by a drastic loss of activity. In several cases, highly promiscuous enzymes were generated.47,49–51
The following Sections 3–8 each focus on one group of currently known chorismate-converting enzymes. Due to the diversity of three-dimensional structures, reactions and mechanisms, there is not a single clear-cut classification scheme. Taking into account the advances in the field over the last decade(s),1–3,7,8 we decided on the following classification of chorismate-converting enzyme families:
• Enzymes Solely catalysing Pericyclic Reactions (SPR-enzymes, Section 3)
• Enzymes involved in the biosynthesis of Menaquinone, Siderophores, and Tryptophan (MST-enzymes, Section 4)
• (Iso-)chorismate hydrolases (isochorismatases) and type-I chorismatases (Section 5)
• SEPHCHC synthases (Section 6)
• Chorismate dehydratases (Section 7)
• Chorismatases (Section 8)
This classification is based on the type of reaction, and in many cases goes along with similar three-dimensional structures and molecular mechanisms. An exception are SPR enzymes, as this group contains subfamilies with different enzyme folds. Chorismatases represent another exception, as they are characterised by a single enzyme fold (Section 8) but perform fundamentally different product formation after a shared initial reaction step (Sections 5 and 8).
3. Enzymes solely catalysing pericyclic reactions: (iso)chorismate mutases and lyases
Although the number of known enzyme families catalysing pericyclic reactions has increased over the last decade, this type of enzymatic reaction remains uncommon.52 (Iso)chorismate mutases, chorismate lyases, and isochorismate-pyruvate lyases solely catalyse pericyclic reactions; in the following, these are referred to as the SPR enzyme family (Solely catalysing Pericyclic Reactions).8,21,29,53 Members of this family are widespread in bacteria, plants, and fungi, and provide important intermediates for the biosynthesis of key primary metabolites such as tyrosine, phenylalanine (chorismate mutases), and ubiquinone (chorismate lyases) (Fig. 2).9,10,34 Beside the primary metabolites, SPR enzymes catalyse the formation of salicylate, and other bioactive secondary metabolites such as pyochelin (isochorismate-pyruvate lyases).8,12 All members of the SPR family share two unique features: (i) no intermediates are formed during the reaction, and (ii) the active site provides an ‘ideal’ environment for product formation without an immediate catalytic contribution.8,21,29(Iso)chorismate mutases convert (iso)chorismate (2/1) into (iso)prephenate (9/5) by a [3,3]-sigmatropic rearrangement (Claisen rearrangement), analogous to the non-enzymatic reactions (Scheme 1).54 The formation of isoprephenate (9) is so far only predicted based on the detection of the product in vivo;55 therefore, it is not clear if dedicated isochorismate mutases exist. Studies on the biosynthesis of the frequently used antibiotic chloramphenicol (22) indicate that one intermediate towards the route to the final product is 4-amino-4-deoxyprephenate (23) provided by a chorismate mutase-like enzyme (ADC mutase),56,57 experimental proof is to our knowledge outstanding. (Iso)chorismate-(pyruvate) lyases catalyse the aromatisation of (iso)chorismate to 4-HBA (6) and salicylate (8) via a [1,5]-sigmatropic rearrangement with elimination of pyruvate (7, Scheme 1).18,19,21,53 In addition to the native reaction, isochorismate-pyruvate lyases show a chorismate mutase side activity with the non-physiological substrate chorismate, albeit with a substantially lower reaction rate.58–60 Other studies explain this chorismate mutase side activity with co-purification of a naturely occurring chorismate mutase from the heterologous host E. coli.48 The corresponding side reaction for wild-type chorismate mutases is not known. Additionally, chorismate lyases perform a non-natural, cofactor-independent elimination reaction of ADC (4) yielding 4-aminobenzoate (24, pABA) and pyruvate (7), which is usually catalysed by pyridoxal phosphate dependent para-aminobenzoate synthases (Scheme 1E).61
3.1. Chorismate mutases and isochorismate-pyruvate lyases catalyse different sigmatropic rearrangements in highly similar active sites
For chorismate mutases two different enzyme folds, the AroH-type and the AroQ-type chorismate mutases, are known, both performing the formation of prephenate. Although they share no clear sequence homology (below 20% identity),29,62–64 the catalytic efficiency (kcat/KM) lies within one order of magnitude (0.95 × 105 to 8.00 × 105 M−1 s−1)29,62–64 and seems to be achieved by a highly similar active site architecture.29 The molar enthalpy of the reaction from chorismate to prephenate was determined as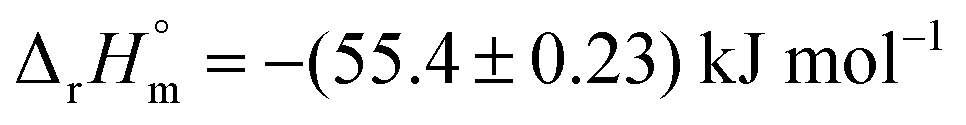 . In context with energy calculations and an equilibrium constant of K ≈ 7 × 109 the reaction can be considered as irreversible.65 AroH-type chorismate mutases, such as the chorismate mutase from B. subtilis, show a pseudo-α/β-barrel structure with a three-domain architecture (Fig. 3D). This results in three active sites at the interfaces between the single domains (Fig. 4D).29 AroQ-type chorismate mutases, e.g. the E. coli chorismate mutase,62–64 share an all α-helical enzyme fold with isochorismate-pyruvate lyases along with a low sequence homology of roughly 20% (Fig. 3A and B). The active sites between the C- and the N-terminus of the single α-helical strands are highly similar (Fig. 4A and B).62–64,66,67
. In context with energy calculations and an equilibrium constant of K ≈ 7 × 109 the reaction can be considered as irreversible.65 AroH-type chorismate mutases, such as the chorismate mutase from B. subtilis, show a pseudo-α/β-barrel structure with a three-domain architecture (Fig. 3D). This results in three active sites at the interfaces between the single domains (Fig. 4D).29 AroQ-type chorismate mutases, e.g. the E. coli chorismate mutase,62–64 share an all α-helical enzyme fold with isochorismate-pyruvate lyases along with a low sequence homology of roughly 20% (Fig. 3A and B). The active sites between the C- and the N-terminus of the single α-helical strands are highly similar (Fig. 4A and B).62–64,66,67
In SPR enzymes the chemical structure of the product formed is controlled by an intricate network of mainly positively and negatively charged amino acid residues in the active site. These residues form an electrostatic gradient over the active site that promotes product formation.19,21,29,43,59 The active site residues involved in development of the electrostatic gradient are highly conserved: among them are two positively charged residues (2 Arg in AroH-chorismate mutases, Arg and Lys in AroQ-chorismate mutases and isochorismate-pyruvate lyases) that coordinate the C2′-methylene group of the enoyl side chain over C1 of the cyclohexadiene ring by electrostatic interactions with the carboxyl group of the enoyl side chain (Fig. 4A, B, and D).19,21 Rearrangement yields prephenate (5) when the not-hybridised p-orbitals of the sp2 carbons overlap, and subsequently the new C–C bond is formed. In a concerted but highly asynchronous reaction, the C–O bond between C3 of the cyclohexadiene ring and the enol ether is broken (Scheme 1A).19 Formation of salicylate (8) from isochorismate (2) is catalysed in a similar fashion to prephenate (5) formation: here, instead of a C–C bond formation, a hydride shift from the sp2 carbon at C2 of the cyclohexadiene ring to the anti-parallel not-hybridised p-orbitals of the sp2 carbon at the C2′-methylene group of the enoyl side chain occurs. In a concerted, asynchronous reaction, the C–O bond between C3 of the cyclohexadiene ring and the enol ether is broken during pyruvate formation and aromatisation yielding salicylate (Scheme 1D).8,21,59,66
In chorismate mutases the C4-hydroxyl group of the cyclohexadiene ring is additionally stabilised by a highly conserved glutamate residue, while isochorismate-pyruvate lyases lack this residue. The resulting lower density of the electrostatic network in isochorismate-pyruvate lyases is assumed to be the reason for the promiscuity of this subfamily in contrast to chorismate mutases (Fig. 4A and B). The importance of the three amino acid residues for catalysis was highlighted by mutagenetic experiments with isosteric amino acid exchanges which resulted in inactive proteins or substantially lower activity.68,69 The pericyclic nature of the isochorismate-pyruvate lyase reaction was shown for PchB by deuterium labelling and subsequent NMR analysis as well as molecular dynamics (MD) simulations.21 The formation of prephenate (5) starting from the non-native substrate chorismate (1) is catalysed via a comparable conformation, as shown for the native substrate isochorismate (Scheme 2D).58,67
3.2. The pericyclic nature of the reaction and the conservation of the active site make chorismate mutases an ideal model system to study enzymatic reactions
Based on the wealth of experimental data, including three-dimensional structures, site-directed mutagenesis, and labelling studies, the pericyclic nature of the chorismate mutase reaction provides an almost ideal system to study this rare enzymatic reaction type theoretically.18,19,21,25,26,29,30,42,54,70 Such studies are based on QM/MM calculations that often compare the sigmatropic rearrangements of chorismate (1) in solution, the gas phase, and the enzymatic environment.22,25,26,30 Depending on the models used for these calculations, different states along the reaction trajectory are recognised as being important for product formation, e.g. the near attack conformation, the transition state, and the pseudo-diaxial conformation of chorismate in the active site.25,29,30,54,71–73 The contribution of these effects has been actively debated for the last two decades. Recent studies support the assumption that the electrostatic gradient, spanned all over the active site, represents the determining factor for catalysis.29,54,74,75Recently, the detailed theoretical and experimental data available for chorismate mutases enabled Russ et al. to succeed in the rational design of artificial enzyme variants with nature-like catalytic activity in vivo.76 These active chorismate mutase variants have remarkable sequence diversity and were predicted by a sequence-based approach using evolutionary data. The results indicate that such statistical models may be of general usefulness for the design of artificial proteins with novel chemical activities.76
3.3. Chorismate lyases have a unique enzyme fold and catalyse a [1,5]-sigmatropic rearrangement
Chorismate lyases such as UbiC from E. coli catalyse the [1,5]-sigmatropic elimination reaction of chorismate (1) yielding pyruvate (7) and 4-HBA (6, Scheme 1B).43 The chorismate lyase from M. tuberculosis is structurally conserved despite 12% sequence homology.77 Chorismate lyases are an exception among chorismate-converting enzymes, since they are known to suffer a strong and competitive product inhibition, that is supposed to stabilise the protein fold.78 The three-dimensional structure has an anti-parallel six-stranded β-sheet covered by two wing-shaped helix-turn-helix motifs forming the active site (Fig. 3C). In addition, these two ‘wings’ are expected to be involved in product release. Another unique feature is the presence of distinct substrate and product binding sites in chorismate lyases.43,53 In contrast to isochorismate-pyruvate lyases, chorismate lyases are specific for their native reaction.43The molecular mechanism is assumed to proceed analogous to the one described for isochorismate-pyruvate lyases. Chorismate (1) is transferred from the substrate to the product binding site, in which the enolpyruvyl tail sterically hinders the stabilisation of chorismate (Fig. 4C). The C2′-methylene group of the enoyl side chain is coordinated over C5 of the cyclohexadiene in a near attack conformation that develops in a pseudo-diaxial chair-like transition state.43,53 A hydride shift from the sp2 carbon at C4 of the cyclohexadiene ring to the anti-parallel not-hybridised p-orbitals of the sp2 carbon at the C2′-methylene group of the enoyl side chain causes a C–O bond cleavage at C3. This results in the formation of pyruvate (7), as well as aromatisation yielding 4-HBA (6, Scheme 1B).43 The same overall reaction is also catalysed by type-III/IV chorismatases (see Section 8).
4. MST enzymes perform nucleophilic substitutions and lyase reactions in one enzyme fold
The enzymes involved in the biosynthesis of Menaquinone, Siderophores, and Tryptophan (MST enzyme family) catalyse the conversion of chorismate (1) into either isochorismate (2), ADIC (3), ADC (4), anthranilate (10), or salicylate (8, Scheme 2).7,8 Anthranilate synthases and ADC synthases, the most abundant MST enzymes, are involved in important pathways of primary metabolism.4,48,79 Salicylate synthases and isochorismate synthases play key roles in the biosynthesis of the electron carrier menaquinone and secondary metabolites, such as the siderophore enterobactin (13).80,81 The plant hormone salicylate plays a key role in plants’ defence against various threats.82–84 Only two ADIC synthases are known to date. This subfamily of MST enzymes is involved in the biosynthesis of phenazines that were shown to have potent antibiotic properties.85 All enzymes of the MST family perform a regio- and stereoselective nucleophilic substitution (SN2′′) resulting in an isomerisation and/or an amination depending on the enzyme and the nucleophile. In the case of anthranilate synthase and salicylate synthase, an additional lyase reaction is catalysed leading to the corresponding aromatised products with loss of pyruvate (7).7,8 Whether the nucleophilic substitution reaction of isochorismate synthases is reversible or irreversible has been controversially debated for decades, based on contradicting experimental results from studies on the isochorismate synthases EntC and MenF.9,864.1. An homologous three-dimensional structure with a highly conserved active site
The overall structure of MST enzymes is dominated by two orthogonal β-sheets decorated with α-helices and loops that determine the final α/β/β/α-fold (Fig. 3G). The active site is located at the C-terminal part of the enzyme structure, embedded between the β-sheets. MST enzymes feature a sophisticated active site with contribution of at least 16 amino acid residues. Nine of these are conserved over all MST enzymes. Three residues slightly differ in their chemical properties, e.g. steric hindrance (Thr vs. Ser), acidic instead of polar (Asn vs. Asp/Glu), or different aromatic side chains (Trp/Phe vs. Tyr). The remaining four amino acids are characteristic for the individual MST subfamilies.79–81,87–90 In addition to the substrate chorismate, a magnesium ion is bound in the active site via four acidic amino acid residues and the substrate's C1 carboxyl group (Fig. 4G).7,91 A recent study by Meneely et al. established the importance of this magnesium ion for catalysis in isochorismate synthases and salicylate synthases.90–94 This is most likely of global impact for MST enzymes, especially for lyase-active enzymes in which the magnesium ion promotes the progress on the reaction trajectory by favouring the formation of reverse protonation states at the catalytic residues glutamate and lysine. This is highlighted by the fact that lyase-deficient isochorismate synthases are inhibited by high concentrations of magnesium, while lyase-active salicylate synthases are not.91 How exactly the formation of such a variety of products is accomplished in a single enzyme fold with an almost conserved active site is still a focus of research on chorismate-converting enzymes (see Section 8 to 10).4.2. MST enzymes carry out regio- and stereoselective nucleophilic substitutions
MST enzymes catalyse at least one SN2-type nucleophilic substitution utilising an acid–base mechanism.7,8,48 The substrate chorismate is protonated at the C4-hydroxyl group while an activated nucleophile (water or ammonia) attacks in a si-fashion at C2 of the cyclohexadiene ring. This results in the formation of a diol/hydroxylamine-carbanion intermediate.7,48 In ADC synthases, the nature and genesis of this intermediate is discussed to be more diverse: ADC synthases such as PabB-II from S. maltophilia use ADIC (3) as an intermediate;95 for other representatives of ADC synthases such as PabB-I from E. coli experimental data indicate an enzyme bound intermediate between the catalytic lysine residue and the chorismate C2 position (Scheme 2C and D).93 Activation of the C4-hydroxyl group at the cyclohexadiene ring is catalysed by a conserved glutamate via protonation and thus the formation of water as the leaving group. The corresponding activation of the nucleophile is achieved via either a basic (Lys) or polar (Asn, Gln) residue. Basic residues are found in MST subfamilies where water serves as a nucleophile (isochorismate synthases, salicylate synthases). Polar residues are found in MST subfamilies where ammonia serves as a nucleophile (Asn – ADIC synthases; Gln – ADC synthases, anthranilate synthases). Water leaves the molecule and causes a shift of the cyclohexadiene ring C–C double bonds and a formal inversion of the stereochemistry regarding the respective hydroxyl or amino moieties attached to C2 and C4 (Scheme 2) of the product compared to the substrate.7,8,48,92–94 In the case of ADC synthases, an alternative SN2-type substitution is catalysed where ammonia attacks at the C4 position of the intermediate described above and causes a retention of the stereochemistry relative to chorismate (1) (Scheme 2C). Currently, the molecular mechanism of ADC synthases remains poorly understood and highly debated due to the low quality of available structural data and contradicting results of studies on the molecular mechanism.4,87,954.3. Enigmatic distinction between lyase activity and lyase deficiency in MST enzymes
The lyase reaction of salicylate and anthranilate synthases is performed in an ordered-sequential mechanism. The long-suggested pericyclic nature of the reaction has been called into doubt by Ziebart and Toney as well as by Culbertson et al. who argue for an acid–base mechanism.48,96 In this case, the active site would promote a conformation of the intermediate isochorismate (2)/ADIC (3) that enables elimination of the enolpyruvyl side chain at C3 and aromatisation of the cyclohexadiene ring (Scheme 2E).8,48,81,91,96 So far, no clear hits for residues that determine lyase activity or deficiency could be obtained.7,48,92,96 A more detailed picture of these studies is given in the recent review by Shelton and Lamb on MST enzymes.75. Isochorismatases and type-I chorismatases (chorismate hydrolases): a typical hydrolysis reaction
Isochorismatases catalyse the hydrolysis of isochorismate (2) to 2,3-trans-dihydroxycyclohexa-4,6-diene-1-carboxylate (25, 2,3-trans-CHD) and pyruvate (7), as well as the hydrolysis of the corresponding amino derivative ADIC (3) yielding 2,3-trans-dihydro-3-hydroxyanthranilate (26, 2,3-trans-CHA).44,97 Beside their physiological reaction, isochorismatases accept chorismate (1) and/or ADC (4) as non-native substrates with substantially lower catalytic efficiency (difference: 104).44,98,99Chorismatases are one of the most recently discovered families of chorismate-converting enzymes. In total, four chorismatase subfamilies catalyse the conversion of chorismate (1) into (dihydro)hydroxybenzoates and pyruvate (7, Scheme 3A).37,50 Type-I chorismatases such as FkbO from S. hygroscopicus subsp. ascomyceticus catalyse the conversion of chorismate (1) into 3,4-trans-dihydroxycyclohexa-1,5-diene-1-carboxylate (27, 3,4-trans-CHD) and pyruvate (7), but do not accept isochorismate (2) as a substrate (Scheme 3B).27,47,100
5.1. A superfamily determining fold and a hydrophobic active site pocket
Isochorismatases have the characteristic α/β-fold of the isochorismatase hydrolase superfamily but lack the catalytic triad that is common to most members of this superfamily.44,98,99,101 The structure is characterised by a central six-stranded β-sheet that is covered on one side by three α-helices and a loop; this also defines the active site. On the opposite side, usually one large α-helix is spanned over the central sheet (Fig. 3F).44,98,99,101 In the active site, isochorismatases feature a hydrophobic pocket stabilising the non-substituted side of the cyclohexadiene ring (Ile, Val, Leu, 2× Phe, 2× Tyr, Trp). A positively charged (Arg) and a polar (Gln) residue coordinate the carboxyl group at C1 while the enolpyruvyl carboxylate is held in place by backbone interactions (Fig. 4F).44,98,99 The presence of an aliphatic amino acid residue (with a variety of side chains resulting in different steric hindrance) is assumed to be responsible for the extent of substrate promiscuity of isochorismatases, as it is the only variable residue across the active site.44,98,99 The chorismatase enzyme fold is unrelated and is discussed in Section 8.1.47,1005.2. An uncommon vinyl ether hydrolysis
Isochorismatases and type-I chorismatases perform their reactions with fundamentally different enzyme folds and active site architectures that are highly conserved within the respective enzyme (sub)family.44,47,98–100 In both suggested mechanisms, an acid protonates the C2′-methylene moiety of the enoyl side chain with development of a carbocation/oxocarbonium ion at the C3′ position. This is subsequently nucleophilically attacked by an activated water molecule in order to form an unstable hemiketal intermediate. This hemiketal decomposes spontaneously into either 3,4-trans-CHD (27) or 2,3-trans-CHD/CHA (25/26) and pyruvate depending on the substrate and the enzyme (Scheme 3).44,47,97 3,4-trans-CHA (28) has also been reported as a product accessible from this hydrolysis in vivo.316. SEPHCHC synthases: a thiamine diphosphate dependent C–C coupling reaction
In contrast to other chorismate-converting enzymes which provide rearranged or cleaved products starting from (iso)chorismate or the corresponding amino derivatives, 2-succinyl-5-enolpyruvyl-6-hydroxycyclohex-3-ene-1-carboxylic acid (29, SEPHCHC) synthases, such as MenD from E. coli, catalyse a C–C coupling reaction of isochorismate (2) and 2-oxoglutarate (30, α-ketoglutarate) yielding SEPHCHC, which can be described as a Stetter-like 1,4-conjugate addition.45 The reaction product 29 is an intermediate in the biosynthesis of menaquinone (vitamin K), an essential electron carrier during anaerobic growth in facultative anaerobic bacteria, e.g. E. coli.6,45 SEPHCHC synthases require thiamine diphosphate (ThDP) as a cofactor and elucidation of different three-dimensional structures has indicated that MenD enzymes are magnesium (as in MST enzymes Section 4) or manganese dependent.91,102,103 Product formation is suggested to proceed via two sequential half-reactions including two covalent intermediates (Scheme 4).104,1056.1. SEPHCHC synthases exhibit a typical ThDP-dependent enzyme fold
The three-dimensional structure of SEPHCHC synthases displays a three-domain architecture characteristic for ThDP-dependent enzymes. Domains I and III share the same topology featuring a central six-stranded β-sheet sandwiched between α-helices; domain II shows a five-stranded β-sheet flanked by six α-helices. The ThDP binding site, as well as the active site, is formed by two subunits of a homodimer at the interface, with contribution of the domains I and III of the different monomers (Fig. 3I).102,103,105 Recently, the function of domain II was elucidated by identification of an allosteric binding site for 1,4-dihydroxy-2-naphthoic acid, a downstream metabolite of menaquinone biosynthesis.106,107In addition to mostly hydrophobic amino acids contributing to the active site, usually five basic amino acid residues provide a suitable chemical environment to promote 1,4-conjugate addition and stabilisation of the product.104,105,107–109 The most important residues that contribute to the process are: (i) a highly conserved glutamate activating ThDP, (ii) two conserved arginines that are suggested to be crucial for substrate binding and catalysis, and (iii) two polar amino acids (Gln and Ser) that finally enable product formation (Fig. 4I).103,105,107–109
6.2. Impressive reaction sequence of the Stetter-like 1,4-conjugate addition of SEPHCHC synthases
In SEPHCHC synthases, isochorismate (2) and 2-oxoglutarate (30) are ThDP-dependently linked in two sequential half-reactions with metastable intermediates (Scheme 4).105 The catalytic cycle of MenD requires as an initial step activation of the cofactor ThDP. The 4-aminopyrimidine ring of ThDP is transformed to the corresponding pyrimidinium and imino tautomers stabilised by the protein environment. These tautomers contribute to the formation of a highly nucleophilic ylide species of the ThDP thiazole ring. C2 of the thiazole ring nucleophilically attacks 2-oxoglutarate to form the post-decarboxylation intermediate I. Mutagenesis as well as structural and kinetic studies indicate that intermediate I is solely present in its carbanion or the protonated C2α-hydroxyalkyl derivative form. This is in contrast to other ThDP-dependent enzymes where the enamine form can be detected as well.102–105,107–109 ThDP and the C2α-decarboxylated 2-oxoglutarate are proposed to be connected in a unique rigid conformation with a defined stereochemistry by two conserved arginine residues keeping intermediate I in the proper orientation for nucleophilic attack at isochorismate, which proceeds with formation of a novel C–C single bond, culminating in intermediate II.105,107–109 Finally, the interplay of two polar amino acids (Gln and Ser), the isochorismate moiety, and the C2α-hydroxyl group of intermediate II, as well as the aminopyrimidine ring of ThDP, in a complex hydrogen-bond network enables product formation. The aminopyrimidine ring of ThDP abstracts a proton from the C2α-hydroxyl group that induces bond breakage between C2 of the ThDP thiazole ring and C2α of intermediate II, leading to regeneration of the cofactor ThDP and formation of the product SEPHCHC (29, Scheme 4).103–105,107–1097. Chorismate dehydratases provide a formal syn-elimination of water
Chorismate dehydratases are the most recent addition to the chorismate-converting enzyme superfamily.46,110 MqnA catalyses the first step of an alternative biosynthetic pathway to menaquinone via aminofutalosine. Genome network analyses indicate that chorismate dehydratases are involved in the biosynthesis of further natural products.6 The only structurally and mechanistically characterised member to date is MqnA from D. radiodurans; the formation of the product 3-enoylpyruvyl benzoate (31) from chorismate was first shown for MqnA from Thermus thermophilus HB8.46,1107.1. Alternative E1cb mechanism of chorismate dehydratases
MqnA follows a variation of an E1cb mechanism in which substrate-mediated deprotonation is slower than loss of water.110 The structure of MqnA was solved with and without 3-HBA (32) as a ligand and product mimic. Detailed information about the overall three-dimensional fold was recently presented as protein data base entries (Fig. 3H).46 The trans periplanar orientation of the substrate chorismate prevents an E2 mechanism. However, a typical E1 mechanism would require activation of the leaving group, which is only likely under acidic or neutral active site conditions. Such conditions were ruled out by the pH profile of MqnA. The putative molecular mechanism of chorismate dehydration starts with an initial slow deprotonation at C3 of the cyclohexadiene ring with formation of a carbanion. The carboxylate of the enolpyruvyl tail is suggested to act as the base for deprotonation, as there is no acidic or basic amino acid residue found in the active site with bound 3-HBA that could assume this role. The carbanion is suggested to be stabilised via delocalisation over the C–C double bonds of the cyclohexadiene ring and the carboxylate at C1. The C4-hydroxyl moiety of the cyclohexadiene ring serves as the leaving group and exits the molecule relatively quickly compared to the other reaction steps (Scheme 5).46 The backbone amide group of a serine residue is assumed to contribute via stabilisation of the leaving group (Fig. 4H).468. Chorismatases: one fold, two mechanisms, three products, four subfamilies
As noted in Section 5 chorismatases can be classified into several subfamilies. In addition to the hydrolysis reaction of type-I chorismatases, type-II, -III, and -IV chorismatases perform a lyase reaction leading to aromatisation of chorismate with release of pyruvate. Type-II chorismatases, such as Hyg5 from S. hygroscopicus subsp. hygroscopicus, catalyse the formation of 3-HBA (32).37,47,111 Type-III chorismatases, such as XanB2 from X. campestris pv. campestris, yield 3-HBA (32) and 4-HBA (6).112–114 Type-IV chorismatases, such as SmCH-IV from S. maltophilia, produce exclusively 4-HBA (6) from chorismate without competitive product inhibition as shown for chorismate lyases, and with comparable catalytic efficiency.50,538.1. A conserved enzyme fold with homology to AroH-chorismate mutases
Remarkably, the different reactions of chorismatases type I, -II, -III, and -IV forming various products are catalysed by a single chorismatase enzyme fold with an average 35% similarity on amino acid basis.47,50 This enzyme fold has a three-domain architecture displaying a pseudo-α/β-barrel structure with homology to the RidA/YjgF superfamily, and, interestingly, AroH-chorismate mutases (Fig. 3D and E). In contrast to the trimeric AroH-chorismate mutases, chorismatases are monomeric containing three similar domains. They have one active site with contribution of amino acid residues from the central and C-terminal domains. A similar architecture, although with the active site located in the center of the molecule, has been described for the cyanuric acid hydrolase AtzD, and coined “Toblerone fold”.115 The location of the active site was elucidated by co-crystallisation of a competitive inhibitor (Fig. 4E): this structure suggests that chorismatases require chorismate to be bound in a pseudo-diaxial conformation for catalysis.47,100,116 Chorismatases coordinate chorismate via a positively charged (Arg) and an aromatic (Tyr) residue at the enolpyruvyl carboxylate. The carboxyl group at C1 is coordinated by an arginine. Further, the substrate is stabilised via π–π interaction of the cyclohexadiene ring and a phenylalanine residue. An acidic residue (Glu) is essential for at least one crucial (de)protonation step during catalysis.47,1008.2. The lyase reaction of chorismatases involves an unexpected arene oxide intermediate
Type-I (FkbO-like) chorismatases catalyse a vinyl ether hydrolysis as described for isochorismatases (see Section 5.2). In contrast to type-I chorismatases, the lyase-active type-II, -III, and -IV chorismatases use a fundamentally different catalysis (Scheme 6). The proposed molecular mechanisms are predominantly based on labelling experiments using selectively labelled substrates and labelled solvents.47,50 A few active site residues in conserved amino acid motifs determine the mechanism and the product(s) formed. A carbocation at the C3′ position is formed through initial protonation at the C2′-methylene group of the enoyl side chain that is common to all four chorismatase subfamilies. In type-II to -IV chorismatases, deprotonation of the C4-hydroxyl group at the cyclohexadiene ring is suggested to be the next crucial step during catalysis, carried out by the same catalytic glutamate used before for protonation. The assumed C4-alcoholate is suggested to perform an intramolecular nucleophilic attack at C3 of the cyclohexadiene ring resulting in the formation of an arene oxide intermediate, with pyruvate as a leaving group. The arene oxide intermediate is opened to yield 3- and/or 4-HBA (32/6) determined by the active site architecture of the respective chorismatase subfamily. The arene oxide opening is proposed to be linked to a concerted NIH shift, characterised by migration of the proton at the position of the product hydroxyl group at the cyclohexadiene ring (C3 – 3-HBA, C4 – 4-HBA) to the ring position where the C–O bond of the arene oxide intermediate was broken. This proton would finally be released during aromatisation of the ring.47,50,117 The fundamental different molecular mechanisms of chorismatases catalysed in highly similar active sites of conserved enzyme folds make chorismatases an interesting target for the theoretical calculation of these reactions.117 These calculations support the proposed molecular mechanisms and suggest further amino acid residues outside of the active site that may play a key role in the selectivity for one distinct molecular mechanism (hydrolysis or arene oxide).24,50,117The respective amino acid residues were also detected as a correlation pair in bioinformatic analysis of the different chorismatase types (see Section 8.3) but ruled out for a role in product formation based on their distance to the active site.50,117
8.3. Three correlating pairs of amino acid residues are responsible for the product outcome of chorismatases
Correlation analysis of a set of (putative) chorismatase protein sequences revealed a pattern of characteristic variable amino acid residues in the active site determining the molecular mechanism catalysed:47,50 in type-I chorismatases two non-polar residues (2× Ala) ensure the correct position of the substrate in the active site for the vinyl ether hydrolysis reaction.50,100 In type-II, -III, and -IV chorismatases, these two residues are substituted by a conserved glycine and a conserved cysteine residue. These substitutions are suggested to change the orientation of chorismate in the active site to enable deprotonation at the C4-hydroxyl group and concomitant formation of the proposed arene oxide intermediate. The product range of type-II, -III, and -IV chorismatases is putatively determined by the electrostatic repulsion and the steric requirements of an active site residue (Gly, Ala, or Cys) adjacent to the C1 carboxylate of the cyclohexadiene ring, and a Glu or Asp residue forming a hydrogen bond with the catalytic glutamate. A combination of Gly and Glu brings the arene oxide intermediate in a position such that the ring opening is selective to provide the meta product 32 likely supported by electrostatic interaction with the Cys residue responsible for mechanistic selectivity. A combination of Ala/Cys and Glu leads to a position of chorismate where the coordinating cysteine residue has no influence on ring opening and the ring opens to the favoured product 4-HBA. Ala and Asp at the respective positions locate chorismate between the two positions in the active site that is linked to a loss of product selectivity.47,50,1178.4. Chorismatases are a starting point for the discovery of novel natural products with rare meta-substituted moieties
Chorismatases are involved in the biosynthesis of a wide range of bioactive natural products. Type-I chorismatases provide the starter unit for the biosynthesis of immunosuppressants such as rapamycin (19, Fig. 2) and ascomycin.37 Type-II and -III chorismatases are involved in the biosynthesis of unusual meta-substituted natural products such as the polyketidic BC325 (a naturally occurring rapamycin derivative), cuevaene A (33), xanthomonadin-dialkyl resorcinol and xanthomonadin I (34), and the terpene brasilicardin (35).37,111–113,118 Only a few other meta-substituted natural products, e.g. 3-formyl-tyrosine and pacidamycin, are known.119,120 Consequently, type-II and -III chorismatases are promising handles to detect novel biosynthetic pathways for compounds with this otherwise rare structural feature. Finally, type-III and -IV chorismatases catalyse the formation of 4-HBA (6) during the biosynthesis of ubiquinones. All studied organisms containing a type-III or -IV chorismatase lack a conventional chorismate lyase in their genome. Therefore, type-III and -IV chorismatases are suggested to represent the starting point of an alternative route to ubiquinone in primary metabolism.50,113 In addition, 4-HBA might serve as starter unit for the biosynthesis of novel arylpolyenes as the studied new type-IV chorismatases are embedded in yet undescribed arylpolyene biosynthetic gene clusters.50 The ability to securely predict the products of an unknown chorismatase is a useful tool for the prediction of novel biosynthetic pathways and was successfully used in the discovery of unantimycins (e.g., 20).38,509. Interchange or permute hypothesis?
As described in the previous sections, chorismate-converting enzymes of one specific (sub)family often share conserved amino acid sequences and are structurally similar (at least) at the active site. The MST enzyme family is an outstanding example of this: Meneely et al. investigated whether product selectivity, the molecular mechanism followed, and the choice of nucleophile in the MST enzyme family are determined by (i) single active site residues, or (ii) a more elaborate interplay of active site residues and electrostatic networks.49 If hypothesis (i) holds true, an interchange of the corresponding enzyme family specific native reactions between different MST enzyme families would be feasible by site-directed mutagenesis (interchange hypothesis). In the case of hypothesis (ii), non-native reactions would be catalysed on a low level after mutagenesis, with reduced catalytic efficiency in addition to the physiological reaction. After site-directed mutagenesis of specific active site residues, the result would be (highly) promiscuous enzymes with a significant loss of product selectivity (permute hypothesis). The interchange or permute hypothesis was tested by attempting to install a lyase activity in lyase-deficient MST enzymes, with the outcome that the permute hypothesis is more likely.49 A final proof for the permute hypothesis is difficult, but most results using site-directed mutagenesis support this finding.4,47,48,80,81,88–90,92,94 Nevertheless, there are a few examples that support the interchange hypothesis as well, e.g. a double mutant of the ADC synthase PabB-I (Lys274Ala/Gln211Lys) that solely produces ADIC under physiological conditions – without any detectable wild-type activity. However, all examples show a significant loss of catalytic activity, thereby raising the question of whether other products would be detected using increased reaction times.48,51,96 In summary, the suggestion that MST enzymes react to variations introduced in the active site with significant promiscuity and loss of product selectivity is reasonable.49This trend is mirrored in other families of chorismate-converting enzymes. Pure interchange approaches in SPR enzymes aimed at establishing a chorismate mutase activity in the isochorismate-pyruvate lyase PchB resulted in promiscuous variants and loss of activity.8,42,67,69,121,122 Similar results were found when site-directed mutagenesis was applied to interchange the different molecular mechanisms of chorismatases: promiscuous variants with median to dramatic loss of catalytic activity were obtained.47,50 Nevertheless, this seemingly undesired loss of selectivity and activity can be seen as a necessary first step on the way to a new activity, which is gradually introduced. Indeed, type-III chorismatases could be regarded as a natively occurring promiscuous variant of lyase-active chorismatases, as this subfamily leads to a mixture of 3-HBA and 4-HBA.47,50,113 The ‘unselectivity’ of type-III chorismatases might thus be an evolutionary snapshot, representing an intermediate step during evolutionary adaption of type-IV towards type-II chorismatases or vice versa. A similar assumption was made for the evolution from anthranilate synthases acting in the primary metabolism to isochorismate synthases acting in the secondary metabolism by exchange of two amino acids instead of gene duplication.51
In conclusion, the interchange experiments performed for different families of chorismate-converting enzymes suggest that distinct chorismate-converting enzyme subfamilies have evolved to be selective for their native reaction. Regarding the reactivity of chorismate and isochorismate, and the number of reactions possible, this requires a precise control of substrate binding and reaction. Therefore, it is not overly surprising that the evolution from one activity to another cannot be easily copied with a few point mutations leading to the same efficiency and selectivity.
10. Recently added enzyme families and mechanistic understanding provide unexplored mechanistic perspectives for chorismate-converting enzymes
As outlined above, chorismate-converting enzymes are a versatile group of enzymes with fascinating and sophisticated molecular mechanisms that selectively lead to a broad range of products. The growing comprehension of the underlying molecular mechanisms in combination with the three-dimensional structures have immediate impact on the broader understanding about the whole group of chorismate-converting enzymes, and might be transferrable to other enzyme families.The recent discovery of chorismatases and chorismate dehydratases added to and shed new light on long held textbook knowledge.37,50,110 Recent studies focused on either the MST or the SPR enzymes have contributed to this process as well.29,49,54,91 In the following, we highlight a few general trends that seem important for the whole range of chorismate-converting enzymes.
10.1. Electrostatic interactions in the active site reflect the reaction's complexity
Two global assumptions can be derived from studies on the structural and mechanistic understanding of chorismate-converting enzymes: (i) a higher number of polar and charged amino acid residues is observed in the active sites of enzyme subfamilies catalysing more intricate mechanisms compared to enzymes with less sophisticated mechanisms of the same family, and (ii) the more complex the molecular mechanism, the higher the degree of promiscuity caused by mutagenesis.It is difficult to clearly define the level of complexity for the different molecular mechanisms. As a guideline we suggest (a) the higher the number of bonds broken and newly formed the more complex the mechanism, and (b) the lower the number of products yielded the more complex the mechanism, particularly when a large number of products can theoretically be formed. In our opinion, these criteria allow to some extent a comparison of complexity between different chorismate-converting enzyme families. As an example, a pericyclic Claisen rearrangement is considered to be more complex than a lyase reaction (including re-aromatisation), which is again of higher complexity than a hydrolysis. Examples of an increased number of polar and charged residues in the active sites of (iso)chorismate-converting enzymes catalysing such sophisticated molecular mechanisms are summarised in the following.
10.2. The C2/C4-hydroxyl group is important for product formation
From structural data and molecular modelling studies it is likely that the pseudo-diaxial conformation of chorismate is a prerequisite for catalysis in chorismate-converting enzymes.7,18,19,21,29,44,46,47 Consequently, the C2/C4-hydroxyl group of isochorismate/chorismate, respectively, usually contributes to some extent to the product formation (Scheme 7).In chorismatases, the molecular mechanisms can be interpreted to resemble the limits of the C4-hydroxyl group's influence on product formation. In type-I chorismatases catalysing a vinyl ether hydrolysis there is a minor influence that is assumed to be limited to the stabilisation of the catalytically active conformation.47,100 In the lyase-catalysing type-II, -III, and -IV chorismatases the C4-hydroxyl group is essential for product formation as it attacks intramolecularly at the C3 position leading to the proposed arene oxide intermediate.47
This situation is also found in other chorismate-converting enzymes. In isochorismatases, the influence of the C2-hydroxyl group on product formation is comparably low, as is the case for the C4-hydroxyl group in type-I chorismatases.97,99 In SEPHCHC synthases, the influence of the C2-hydroxyl group seems to be minor as well.105,108 In MST enzymes, the C2/C4-hydroxyl group has an immediate influence on product formation as it serves as a leaving group for the nucleophilic substitution while being (re)installed during the same reaction. Its presence is therefore essential but different to the situation in the chorismatase subfamilies.7 The same might be true for chorismate dehydratases: here, the C4-hydroxyl group is the leaving group during elimination of water and subsequent aromatisation.46
The influence of the C2/C4-hydroxyl group, especially in lyase-catalysing chorismatases, indicates a significant importance of this moiety for product formation in (iso)chorismate-converting enzymes: it is conceivable that the C2/C4-hydroxyl group also essentially contributes to the molecular mechanisms of SPR enzymes as well as the lyase-active MST enzymes. This contribution could be based on stereoelectronic effects, e.g. hyperconjugation or the anomeric effect, or the development of an arene oxide intermediate. The latter has been suggested for lyase-catalysing chorismatases, as further outlined in the following.
10.3. Is the arene oxide postulated for chorismatases also a feasible intermediate for other reactions?
The recent discovery of chorismate dehydratases and chorismatases highlights that intramolecular reactions with contribution of acidic and/or basic active site residues are not unusual in chorismate-converting enzymes.46,47 This raises the question of whether the pericyclic reactions in chorismate-converting enzymes are just formally pericyclic, with true pericyclic non-enzymatic counterparts in solution. Several results of studies on enzymatic and non-enzymatic reactions of (iso)chorismate support this hypothesis.The pericyclic reactions catalysed by SPR enzymes, the lyase reaction of MST enzymes, and the reactions catalysed by chorismatases require a pseudo-diaxial conformation of (iso)chorismate, for which the enzymes are assumed to be selective.24,25,29,30,54,71–75,117 In all these cases, the position of the enolpyruvyl tail is essential for product formation and determines the molecular mechanism and the product selectivity in a complex interplay with polar and/or charged amino acid residues in the active site.7,19,21,47,100 Consequently, if the enolpyruvyl side chain is of general importance for product formation and (iso)chorismate has a pseudo-diaxial conformation, the C2/C4-hydroxyl group as its counterpart is likewise of general importance for product formation. Studies with 4-deoxychorismate and 4-O-methylchorismate in solution suggest that an anomeric effect caused by the additional methyl group increases the reaction rate by weakening the C–O bond at C3 of chorismate more than the hydroxyl group usually does (Scheme 7).123 Moreover, Hilvert et al. observed a difference between the theoretical calculations for the pericyclic reactions of (iso)chorismate and the experimental results for the enzymatic but not for the non-enzymatic reactions.18,19,21 Finally, the finding that the reaction proceeds in a highly asynchronous but pericyclic manner seems to be a contradiction in itself. Consequently, these studies might serve as a hint that the C2/C4-hydroxyl group is of general importance for catalysis.18,19,21 Although shown by kinetic isotope effects that the reactions proceed in a concerted fashion,18,19,21 the concerted nature of the reaction does not exclude the formation of an elusive arene oxide intermediate as suggested for type-II, -III, and -IV chorismatases.47,50 In chorismate mutases, an alternative reaction mechanism with an arene oxide intermediate has been suggested before.124 The conserved glutamate residue interacting with the C4-hydroxyl group might serve as the base for deprotonation and an additional cysteine residue would be responsible for the faster opening of the arene oxide to the C4 position. This cysteine residue would be an explanation for the higher catalytic efficiency in AroH-chorismate mutases compared to AroQ-chorismate mutases that lack this residue.29,62
An arene oxide intermediate has been suggested also in the biosynthesis of salicylate.125 This idea can be transferred to other lyase reactions of chorismate, as it was indirectly shown for chorismatases that the formation of 4-HBA proceeds most likely via such an intermediate.47,50 The introduction of deuterium into pyruvate from selectively deuterium-labelled isochorismate and the asynchronous nature of the isochorismate-pyruvate lyase reaction support a pericyclic reaction as well as an arene oxide intermediate.21 Judging from the enzyme structures available, the active site environment of isochorismate-pyruvate lyases, as well as of salicylate synthases and anthranilate synthases, would be suitable to stabilise an arene oxide intermediate. An amino acid residue that could serve as the base is also present in these enzymes.¶![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7,21,67 Moreover, structural data of salicylate and anthranilate synthases show that the enolpyruvyl side chain is embedded in a dense network of hydrogen bonds, keeping this part of the molecule firmly in position during product formation.7,81,96 This rigid network might actually hinder the movement of the side chain over the ring during the lyase reaction, thus making a pericyclic reaction less likely. In chorismatases, the enolpyruvyl tail is suggested to assume a similar position during formation of the arene oxide intermediate.47
7,21,67 Moreover, structural data of salicylate and anthranilate synthases show that the enolpyruvyl side chain is embedded in a dense network of hydrogen bonds, keeping this part of the molecule firmly in position during product formation.7,81,96 This rigid network might actually hinder the movement of the side chain over the ring during the lyase reaction, thus making a pericyclic reaction less likely. In chorismatases, the enolpyruvyl tail is suggested to assume a similar position during formation of the arene oxide intermediate.47
In summary, an alternative reaction mechanism with formation of an arene oxide intermediate for other chorismate-converting enzymes is imaginable but to date elusive. Experimental approaches to support this hypothesis are challenging as the arene oxide intermediate has to be scavenged by strong nucleophiles and/or detected spectroscopically.126
11. The metabolic flux into the different branches starting from (iso)chorismate is directed by allosteric regulation
Chorismate is a metabolic node with many different branches; consequently, in each organism with a shikimate biosynthetic pathway multiple chorismate-converting enzymes are found.1 The metabolic flux into the specific branches plays a key role in biosynthesis of the corresponding (iso)chorismate derivatives. In addition to straightforward mechanisms such as the product inhibition described for chorismate lyases (see SPR enzyme section), an important way to control this flux is allosteric regulation. The binding of an allosteric regulator distant from the active site can cause feedback inhibition or activation of the catalytic activity.127Allosteric regulation of chorismate-converting enzymes was first identified for chorismate mutases and their involvement in the biosynthesis of aromatic amino acids. Accordingly, it was shown that many chorismate mutases from various organisms are allosterically regulated via feedback inhibition by the aromatic amino acids Phe, Tyr, and Trp.128 Additionally, inter-enzyme allostery has been reported for chorismate mutase, e.g. in M. tuberculosis and Corynebacterium glutamicum where Trp-bound 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase binds to the inactive chorismate mutase, with the resulting complex increasing chorismate mutase activity thereby directing additional flux into the biosynthesis of aromatic amino acids.54,129,130
In MST enzymes, allosteric regulation has been described for the glutamine amidotransferase domain of ADC synthases. Here, the formation of the nucleophile ammonia is allosterically activated by chorismate, binding in the active site. This concurs with the need of the nucleophile for catalysis.131 The allosteric activation of the amidotransferase domain ensures that glutamine is only consumed if chorismate is available for conversion into the corresponding amino derivative ADC. Trp abolishes catalysis in anthranilate synthases allosterically by effecting an ammonia channel to the active site without major structural changes.132 Additionally, structural data with bound Trp imply that Trp binding induces changes in the hydrogen-binding patterns of anthranilate synthase hindering the optimal formation of the active site.133
Recently, allosteric regulation of the SEPHCHC synthase MenD from M. tuberculosis was identified to be essential for the metabolic flux of menaquinone biosynthesis.106,107 The catalytic activity of MenD is inhibited by binding of 1,4-dihydroxy-2-naphthoic acid in an ‘arginine cage’ located at domain II of the enzyme. Binding of the inhibitor at the allosteric site causes structural changes that affect the position of a catalytically essential glutamate residue as well as the flexibility of the active site. Nevertheless, a multi-sequence alignment of 35 MenD amino acid residues revealed that the three key arginine residues of the allosteric binding site show low conservation. This indicates that allosteric regulation of MenD by 1,4-dihydroxy-2-naphthoic acid is not a universal feature to direct metabolic flux in menaquinone biosynthesis.106,107
In conclusion, the identification of allosteric control in various chorismate-converting enzymes suggests that future work in this direction might reveal such control to be a widespread feature to direct metabolic flux into the different (iso)chorismate biosynthetic pathways.
12. Chorismate-converting enzymes are important tools for biotechnological and biocatalytic applications
Chiral building blocks are often challenging to access by conventional organic synthesis. Enzymes in general are emerging as biocatalysts for the selective chemoenzymatic synthesis of chiral building blocks in vitro and in vivo.134,135 The metabolic branching point(s) of (iso)chorismate offer access to many chiral and/or aromatic compounds.31 Several of these compounds have been made accessible on gram scale from rationally engineered E. coli strains, based on a growing understanding of metabolic flux of the biosynthetic pathways branching from (iso)chorismate.11,136,137 Microbial production of chemicals in vivo is complemented with the in vitro production of compounds such as isochorismate itself.27In addition to such technical applications, chorismate-converting enzymes harbour a large potential as biocatalysts. An outstanding example among chorismate-converting enzymes are the SEPHCHC synthases. The promiscuity of MenD makes this enzyme a versatile in vitro catalyst for the asymmetric synthesis of numerous 2-hydroxy ketones by variation of the donor and acceptor substrates.138,139 The non-physiological isochorismate derivatives SDHCHC (37) and MDHCHC (38) from the reactions of 2,3-trans-CHD (25) with 2-oxoglutarate or (S)-4-hydroxy-2-oxoglutarate, respectively, are accessible by wild-type MenD in vitro (Fig. 4). Recently, the substrate and product range of wild-type MenD was extended by (5S,6S)-6-amino-2-(3-carboxypropanoyl)-5-hydroxycyclohex-3-ene-1-carboxylate (38, SAHCHC) that was detected in traces from the conversion of 2,3-trans-CHA (26) with 2-oxoglutarate (Fig. 5).31,32,139
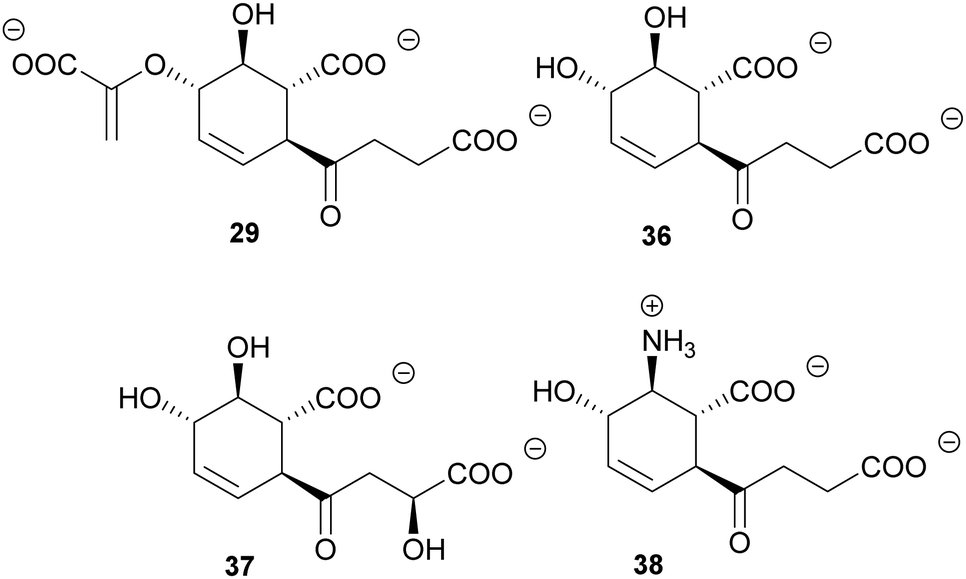 | ||
| Fig. 5 SEPHCHC (29) and derivatives 36–38 from biocatalytic applications of wild-type MenD and variants thereof. | ||
Moreover, Fries et al. reported the preparative in vivo production of SEPHCHC, (iso)SDHCHC, and (iso)SAHCHC using engineered E. coli strains. Rationally designed MenD variants, based on a docking model with ‘1,6-dihydroisochorismate’ as a surrogate intermediate, increased the yields of fermentations and in vitro reactions significantly (6- and 23-fold). The occurrence of some of these rationally designed variants in nature, detected by protein sequence network analysis, suggests that further chorismate-converting enzymes – in particular enzymes acting on the amino derivatives – await discovery.32
13. Is there a hidden chorismate-converting enzyme potential encoded in natural products?
In addition to the chorismate-converting enzymes characterised regarding their three-dimensional structures and molecular mechanisms, natural products provide insight into the cryptic potential of enzymatic novelty by retro-biosynthetic analysis and scavenging of intermediates. In light of this, it is likely that further unprecedented enzyme families will be discovered.32,46,50 Reconsidering Fig. 1, the products accessible from (iso)chorismate (1 and 2) by enzymatic and non-enzymatic reactions are not fully covered for the corresponding amino derivatives ADIC (3) and ADC (4). Identification of such as yet enigmatic enzymes might lead to the amino derivatives being recognised as important metabolic branching points contributing to a much larger biosynthetic diversity, as was proposed for the biosynthesis of stravidins (18), echinosporins (39), and SEPHCHC derivatives (36–38) prepared with variants of MenD.32,39,140 In addition, the elucidation of the tundrenone (40) biosynthetic pathway suggests an undescribed C–C coupling reaction utilising chorismate as a substrate (Fig. 6).141 | ||
| Fig. 6 Natural products that are expected to be biosynthesised via a route containing uncharacterised chorismate-converting enzymes: echinosporins (39) and tundrenone (40) of bacterial origin, the fungal natural products chorismeron (41) and shikimeran B (42), and the plant metabolite glutamyl-isochorismate (43). | ||
Higher organisms provide another putative source of unexplored chorismate-converting enzymes. Consequently, co-cultivation of fungi with different bacteria led to the identification followed by characterisation of structurally unique fungal chorismate metabolites, e.g. chorismeron (41) and shikimeran B (42) (Fig. 5).142,143 Further, an unprecedented chorismate derivative was detected in plants, where salicylate biosynthesis occurs via a three-enzyme cascade in two cell compartments. Glutamyl-isochorismate (43) plays a key role by releasing the final natural product salicylate via a lyase reaction (Fig. 6).82–84
14. Conclusions
The recent advances in the field of chorismate-converting enzymes demonstrate the dynamic nature of this field. Many enigmatic processes require further studies to enable a thorough understanding of such a unique metabolic node. In addition, recent studies on biosynthetic pathways of natural products and bioinformatic analysis directed towards the abundance of recently discovered chorismate-converting enzymes indicate that there is a large and untapped enzymatic and natural product potential. Therefore, we are convinced that this metabolic node will continue its action in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Jodie Johnston (University of Canterbury, NZ), Prof. Georg A. Sprenger and Prof. Jürgen Pleiss (University of Stuttgart, Germany), and Prof. Kai Tittmann (University of Göttingen, Germany) for many scientific and philosophical discussions on chorismate-converting enzymes in the last 10+ years; as well as Dr Jodie Johnston, Prof. Georg A. Sprenger, and Dr Kay Greenfield (Brisbane, Australia) for proofreading of and helpful comments on the manuscript.Notes and references
- F. Dosselaere and J. Vanderleyden, Crit. Rev. Microbiol., 2001, 27, 75–131 CrossRef CAS.
- R. Bentley and E. Haslam, Crit. Rev. Biochem. Mol. Biol., 1990, 25, 307–384 CrossRef CAS.
- H. G. Floss, Nat. Prod. Rep., 1997, 14, 433–452 RSC.
- J. F. Parsons, P. Y. Jensen, A. S. Pachikara, A. J. Howard, E. Eisenstein and J. E. Ladner, Biochemistry, 2002, 41, 2198–2208 CrossRef CAS.
- A. A. Morollo and R. Bauerle, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 9983–9987 CrossRef CAS.
- S. Joshi, D. Fedoseyenko, N. Mahanta, H. Manion, S. Naseem, T. Dairi and T. P. Begley, Curr. Opin. Chem. Biol., 2018, 47, 134–141 CrossRef CAS.
- C. L. Shelton and A. L. Lamb, Trends Biochem. Sci., 2018, 43, 342–357 CrossRef CAS.
- A. L. Lamb, Biochemistry, 2011, 50, 7476–7483 CrossRef CAS.
- R. Meganathan, Vitam. Horm., 2001, 61, 173–218 CrossRef CAS.
- S. Noda and A. Kondo, Trends Biotechnol., 2017, 35, 785–796 CrossRef CAS.
- G. A. Sprenger, Appl. Microbiol. Biotechnol., 2007, 75, 739–749 CrossRef CAS.
- H. Maeda and N. Dudareva, Annu. Rev. Plant Biol., 2012, 63, 73–105 CrossRef CAS.
- C. A. Schenck and H. A. Maeda, Phytochemistry, 2018, 149, 82–102 CrossRef CAS.
- A. Kaiser and E. Leistner, Arch. Biochem. Biophys., 1990, 276, 331–335 CrossRef CAS.
- T. Hiratsuka, K. Furihata, J. Ishikawa, H. Yamashita, N. Itoh, H. Seto and T. Dairi, Science, 2008, 321, 1670–1673 CrossRef CAS.
- S. D. Copley and J. R. Knowles, J. Am. Chem. Soc., 1987, 109, 5008–5013 CrossRef CAS.
- K. M. Mattia and B. Ganem, J. Org. Chem., 1994, 59, 720–728 CrossRef CAS.
- M. S. DeClue, K. K. Baldridge, P. Kast and D. Hilvert, J. Am. Chem. Soc., 2006, 128, 2043–2051 CrossRef CAS.
- S. K. Wright, M. S. DeClue, A. Mandal, L. Lee, O. Wiest, W. W. Cleland and D. Hilvert, J. Am. Chem. Soc., 2005, 127, 12957–12964 CrossRef CAS.
- K. A. Reynolds, K. K. Wallace, S. Handa, M. S. Brown, H. A. McArthur and H. G. Floss, J. Antibiot., 1997, 50, 701–703 CrossRef CAS.
- M. S. DeClue, K. K. Baldridge, D. E. Künzler, P. Kast and D. Hilvert, J. Am. Chem. Soc., 2005, 127, 15002–15003 CrossRef CAS.
- S. Hur and T. C. Bruice, J. Am. Chem. Soc., 2003, 125, 10540–10542 CrossRef CAS.
- S. Hur and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12015–12020 CrossRef CAS.
- Y. Zhang, H. Zhang and Q. Zheng, Chem. – Eur. J., 2019, 25, 1326–1336 CAS.
- K. E. Ranaghan and A. J. Mulholland, Chem. Commun., 2004, 1238–1239 RSC.
- F. Claeyssens, K. E. Ranaghan, F. R. Manby, J. N. Harvey and A. J. Mulholland, Chem. Commun., 2005, 5068–5070 RSC.
- F. Hubrich, M. Müller and J. N. Andexer, J. Biotechnol., 2014, 191, 93–98 CrossRef CAS.
- Y. B. Tewari, A. M. Davis, P. Reddy and R. N. Goldberg, J. Chem. Thermodyn., 2000, 32, 1057–1070 CrossRef CAS.
- D. Burschowsky, A. van Eerde, M. Okvist, A. Kienhöfer, P. Kast, D. Hilvert and U. Krengel, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17516–17521 CrossRef CAS.
- X. Zhang and T. C. Bruice, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18356–18360 CrossRef CAS.
- J. Bongaerts, S. Esser, V. Lorbach, L. Al-Momani, M. A. Müller, D. Franke, C. Grondal, A. Kurutsch, R. Bujnicki, R. Takors, L. Raeven, M. Wubbolts, R. Bovenberg, M. Nieger, M. Schürmann, N. Trachtmann, S. Kozak, G. A. Sprenger and M. Müller, Angew. Chem., Int. Ed., 2011, 50, 7781–7786 CrossRef CAS.
- A. Fries, L. S. Mazzaferro, B. Grüning, P. Bisel, K. Stibal, P. C. F. Buchholz, J. Pleiss, G. A. Sprenger and M. Müller, ChemBioChem, 2019, 20, 1672–1677 CrossRef CAS.
- C. T. Walsh, S. W. Haynes and B. D. Ames, Nat. Prod. Rep., 2012, 29, 37–59 RSC.
- V. Tzin and G. Galili, Mol. Plant, 2010, 3, 956–972 CrossRef CAS.
- F. Gibson and L. M. Jackman, Nature, 1963, 198, 388–389 CrossRef CAS.
- A. R. Knaggs, Nat. Prod. Rep., 2003, 20, 119–136 RSC.
- J. N. Andexer, S. G. Kendrew, M. Nur-e-Alam, O. Lazos, T. A. Foster, A.-S. Zimmermann, T. D. Warneck, D. Suthar, N. J. Coates, F. E. Koehn, J. S. Skotnicki, G. T. Carter, M. A. Gregory, C. J. Martin, S. J. Moss, P. F. Leadlay and B. Wilkinson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4776–4781 CrossRef CAS.
- Y. Shen, F. Sun, L. Zhang, Y. Cheng, H. Zhu, S.-P. Wang, W.-H. Jiao, P. F. Leadlay, Y. Zhou and H.-W. Lin, J. Biol. Chem., 2020, 295, 5509–5518 CrossRef CAS.
- R. Montaser and N. L. Kelleher, ACS Chem. Biol., 2020, 15, 1134–1140 CrossRef CAS.
- P. Moosmann, R. Ueoka, L. Grauso, A. Mangoni, B. I. Morinaka, M. Gugger and J. Piel, Angew. Chem., Int. Ed., 2017, 56, 4987–4990 CrossRef CAS.
- J. P. Torres and E. W. Schmidt, J. Biol. Chem., 2019, 294, 17684–17692 CrossRef.
- J. K. Lassila, J. R. Keeffe, P. Kast and S. L. Mayo, Biochemistry, 2007, 46, 6883–6891 CrossRef CAS.
- N. Smith, A. E. Roitberg, E. Rivera, A. Howard, M. J. Holden, M. Mayhew, S. Kaistha and D. T. Gallagher, Arch. Biochem. Biophys., 2006, 445, 72–80 CrossRef CAS.
- J. F. Parsons, K. Calabrese, E. Eisenstein and J. E. Ladner, Biochemistry, 2003, 42, 5684–5693 CrossRef CAS.
- M. Jiang, Y. Cao, Z.-F. Guo, M. Chen, X. Chen and Z. Guo, Biochemistry, 2007, 46, 10979–10989 CrossRef CAS.
- N. Mahanta, K. A. Hicks, S. Naseem, Y. Zhang, D. Fedoseyenko, S. E. Ealick and T. P. Begley, Biochemistry, 2019, 58, 1837–1840 CrossRef CAS.
- F. Hubrich, P. Juneja, M. Müller, K. Diederichs, W. Welte and J. N. Andexer, J. Am. Chem. Soc., 2015, 137, 11032–11037 CrossRef CAS.
- K. T. Ziebart and M. D. Toney, Biochemistry, 2010, 49, 2851–2859 CrossRef CAS.
- K. M. Meneely, Q. Luo and A. L. Lamb, Arch. Biochem. Biophys., 2013, 539, 70–80 CrossRef CAS.
- M. J. Grüninger, P. C. F. Buchholz, S. Mordhorst, P. Strack, M. Müller, F. Hubrich, J. Pleiss and J. N. Andexer, Org. Biomol. Chem., 2019, 17, 2092–2098 RSC.
- M. G. Plach, P. Löffler, R. Merkl and R. Sterner, Angew. Chem., Int. Ed., 2015, 54, 11270–11274 CrossRef CAS.
- R. Little, F. C. R. Paiva, R. Jenkins, H. Hong, Y. Sun, Y. Demydchuk, M. Samborskyy, M. Tosin, F. J. Leeper, M. V. B. Dias and P. F. Leadlay, Nat. Catal., 2019, 2, 1045–1054 CrossRef CAS.
- D. T. Gallagher, M. Mayhew, M. J. Holden, A. Howard, K.-J. Kim and V. L. Vilker, Proteins: Struct., Funct., Bioinf., 2001, 44, 304–311 CrossRef CAS.
- D. Burschowsky, U. Krengel, E. Uggerud and D. Balcells, FEBS Open Bio, 2017, 7, 789–797 CrossRef CAS.
- L. O. Zamir, A. Nikolakakis, C. A. Bonner and R. A. Jensen, Bioorg. Med. Chem. Lett., 1993, 3, 1441–1446 CrossRef CAS.
- J. He, N. Magarvey, M. Piraee and L. C. Vining, Microbiology, 2001, 147, 2817–2829 CrossRef CAS.
- K. Yanai, N. Sumida, K. Okakura, T. Moriya, M. Watanabe and T. Murakami, Nat. Biotechnol., 2004, 22, 848–855 CrossRef CAS.
- D. E. Künzler, S. Sasso, M. Gamper, D. Hilvert and P. Kast, J. Biol. Chem., 2005, 280, 32827–32834 CrossRef.
- Q. Luo, K. M. Meneely and A. L. Lamb, J. Am. Chem. Soc., 2011, 133, 7229–7233 CrossRef CAS.
- A. Choutko, A. P. Eichenberger, W. F. van Gunsteren and J. Dolenc, Protein Sci., 2013, 22, 809–822 CrossRef CAS.
- B. P. Nichols and J. M. Green, J. Bacteriol., 1992, 174, 5309–5316 CrossRef CAS.
- M. Ökvist, R. Dey, S. Sasso, E. Grahn, P. Kast and U. Krengel, J. Mol. Biol., 2006, 357, 1483–1499 CrossRef.
- R. Qamra, P. Prakash, B. Aruna, S. E. Hasnain and S. C. Mande, Biochemistry, 2006, 45, 6997–7005 CrossRef CAS.
- S.-K. Kim, S. K. Reddy, B. C. Nelson, H. Robinson, P. T. Reddy and J. E. Ladner, FEBS J., 2008, 275, 4824–4835 CrossRef CAS.
- P. Kast, Y. B. Tewari, O. Wiest, D. Hilvert, K. N. Houk and R. N. Goldberg, J. Phys. Chem. B, 1997, 101, 10976–10982 CrossRef CAS.
- J. Olucha, K. M. Meneely and A. L. Lamb, Biochemistry, 2012, 51, 7525–7532 CrossRef CAS.
- Q. Luo, J. Olucha and A. L. Lamb, Biochemistry, 2009, 48, 5239–5245 CrossRef CAS.
- P. Kast, J. D. Hartgerink, M. Asif-Ullah and D. Hilvert, J. Am. Chem. Soc., 1996, 118, 3069–3070 CrossRef CAS.
- D. R. Liu, S. T. Cload, R. M. Pastor and P. G. Schultz, J. Am. Chem. Soc., 1996, 118, 1789–1790 CrossRef CAS.
- S. Martí, J. Andrés, V. Moliner, E. Silla, I. Tuñón and J. Bertrán, Interdiscip. Sci.: Comput. Life Sci., 2010, 2, 115–131 CrossRef.
- S. Hur and T. C. Bruice, J. Am. Chem. Soc., 2003, 125, 5964–5972 CrossRef CAS.
- A. Kienhöfer, P. Kast and D. Hilvert, J. Am. Chem. Soc., 2003, 125, 3206–3207 CrossRef.
- S. D. Copley and J. R. Knowles, J. Am. Chem. Soc., 1985, 107, 5306–5308 CrossRef CAS.
- M. Freindorf, Y. Tao, D. Sethio, D. Cremer and E. Kraka, Mol. Phys., 2019, 117, 1172–1192 CrossRef CAS.
- L. Xie, M. Yang and Z.-N. Chen, Catal. Sci. Technol., 2019, 9, 957–965 RSC.
- W. P. Russ, M. Figliuzzi, C. Stocker, P. Barrat-Charlaix, M. Socolich, P. Kast, D. Hilvert, R. Monasson, S. Cocco, M. Weigt and R. Ranganathan, Science, 2020, 369, 440–445 CAS.
- G. Stadthagen, J. Korduláková, R. Griffin, P. Constant, I. Bottová, N. Barilone, B. Gicquel, M. Daffé and M. Jackson, J. Biol. Chem., 2005, 280, 40699–40706 CrossRef CAS.
- M. J. Holden, M. P. Mayhew, D. T. Gallagher and V. L. Vilker, Biochim. Biophys. Acta, 2002, 1594, 160–167 CrossRef CAS.
- T. Knöchel, A. Ivens, G. Hester, A. Gonzalez, R. Bauerle, M. Wilmanns, K. Kirschner and J. N. Jansonius, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9479–9484 CrossRef.
- S. Sridharan, N. Howard, O. Kerbarh, M. Błaszczyk, C. Abell and T. L. Blundell, J. Mol. Biol., 2010, 397, 290–300 CrossRef CAS.
- J. Zwahlen, S. Kolappan, R. Zhou, C. Kisker and P. J. Tonge, Biochemistry, 2007, 46, 954–964 CrossRef CAS.
- C. K. Holland, C. S. Westfall, J. E. Schaffer, A. D. Santiago, C. Zubieta, S. Alvarez and J. M. Jez, J. Biol. Chem., 2019, 294, 16855–16864 CrossRef CAS.
- D. Rekhter, D. Lüdke, Y. Ding, K. Feussner, K. Zienkiewicz, V. Lipka, M. Wiermer, Y. Zhang and I. Feussner, Science, 2019, 365, 498–502 CrossRef CAS.
- M. P. Torrens-Spence, A. Bobokalonova, V. Carballo, C. M. Glinkerman, T. Pluskal, A. Shen and J.-K. Weng, Mol. Plant, 2019, 12, 1577–1586 CrossRef CAS.
- M. McDonald, D. V. Mavrodi, L. S. Thomashow and H. G. Floss, J. Am. Chem. Soc., 2001, 123, 9459–9460 CrossRef CAS.
- K. Buss, R. Müller, C. Dahm, N. Gaitatzis, E. Skrzypczak-Pietraszek, S. Lohmann, M. Gassen and E. Leistner, Biochim. Biophys. Acta, 2001, 1522, 151–157 CrossRef CAS.
- A. K. Bera, V. Atanasova, A. Dhanda, J. E. Ladner and J. F. Parsons, Biochemistry, 2012, 51, 10208–10217 CrossRef CAS.
- Q.-A. Li, D. V. Mavrodi, L. S. Thomashow, M. Roessle and W. Blankenfeldt, J. Biol. Chem., 2011, 286, 18213–18221 CrossRef CAS.
- K. M. Meneely, Q. Luo, P. Dhar and A. L. Lamb, Arch. Biochem. Biophys., 2013, 538, 49–56 CrossRef CAS.
- S. Kolappan, J. Zwahlen, R. Zhou, J. J. Truglio, P. J. Tonge and C. Kisker, Biochemistry, 2007, 46, 946–953 CrossRef CAS.
- K. M. Meneely, J. A. Sundlov, A. M. Gulick, G. R. Moran and A. L. Lamb, J. Am. Chem. Soc., 2016, 138, 9277–9293 CrossRef CAS.
- O. Kerbarh, A. Ciulli, D. Y. Chirgadze, T. L. Blundell and C. Abell, ChemBioChem, 2007, 8, 622–624 CrossRef CAS.
- O. Kerbarh, E. M. M. Bulloch, R. J. Payne, T. Sahr, F. Rébeillé and C. Abell, Biochem. Soc. Trans., 2005, 33, 763–766 CrossRef CAS.
- Z. He, K. D. Stigers Lavoie, P. A. Bartlett and M. D. Toney, J. Am. Chem. Soc., 2004, 126, 2378–2385 CrossRef CAS.
- H. S. Schadt, S. Schadt, F. Oldach and R. D. Süssmuth, J. Am. Chem. Soc., 2009, 131, 3481–3483 CrossRef CAS.
- J. E. Culbertson, D. Hee Chung, K. T. Ziebart, E. Espiritu and M. D. Toney, Biochemistry, 2015, 54, 2372–2384 CrossRef CAS.
- F. Rusnak, J. Liu, N. Quinn, G. A. Berchtold and C. T. Walsh, Biochemistry, 1990, 29, 1425–1435 CrossRef CAS.
- S. Liu, C. Zhang, N. Li, B. Niu, M. Liu, X. Liu, T. Wei, D. Zhu, Y. Huang, S. Xu and L. Gu, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 1329–1338 CrossRef CAS.
- E. J. Drake, D. A. Nicolai and A. M. Gulick, Chem. Biol., 2006, 13, 409–419 CrossRef CAS.
- P. Juneja, F. Hubrich, K. Diederichs, W. Welte and J. N. Andexer, J. Mol. Biol., 2014, 426, 105–115 CrossRef CAS.
- J. Du, T. Deng and Q. Ma, Biochem. Biophys. Res. Commun., 2017, 490, 827–833 CrossRef CAS.
- A. Dawson, P. K. Fyfe and W. N. Hunter, J. Mol. Biol., 2008, 384, 1353–1368 CrossRef CAS.
- A. Dawson, M. Chen, P. K. Fyfe, Z. Guo and W. N. Hunter, J. Mol. Biol., 2010, 401, 253–264 CrossRef CAS.
- H. Song, C. Dong, M. Qin, Y. Chen, Y. Sun, J. Liu, W. Chan and Z. Guo, J. Am. Chem. Soc., 2016, 138, 7244–7247 CrossRef CAS.
- E. N. M. Jirgis, G. Bashiri, E. M. M. Bulloch, J. M. Johnston and E. N. Baker, Structure, 2016, 24, 1167–1177 CrossRef CAS.
- J. M. Johnston and E. M. Bulloch, Curr. Opin. Struct. Biol., 2020, 65, 33–41 CrossRef CAS.
- G. Bashiri, L. V. Nigon, E. N. M. Jirgis, N. A. T. Ho, T. Stanborough, S. S. Dawes, E. N. Baker, E. M. M. Bulloch and J. M. Johnston, J. Biol. Chem., 2020, 295, 3759–3770 CrossRef CAS.
- M. Qin, H. Song, X. Dai, Y. Chen and Z. Guo, Biochem. J., 2018, 475, 3651–3667 CrossRef CAS.
- M. Qin, H. Song, X. Dai, C.-K. Chan, W. Chan and Z. Guo, ChemBioChem, 2018, 19, 1514–1522 CrossRef CAS.
- N. Mahanta, D. Fedoseyenko, T. Dairi and T. P. Begley, J. Am. Chem. Soc., 2013, 135, 15318–15321 CrossRef CAS.
- Y. Jiang, H. Wang, C. Lu, Y. Ding, Y. Li and Y. Shen, ChemBioChem, 2013, 14, 1468–1475 CrossRef CAS.
- T. A. Schöner, S. W. Fuchs, B. Reinhold-Hurek and H. B. Bode, PLoS One, 2014, 9, e90922 CrossRef.
- L. Zhou, J. Wang, J. Wang, A. Poplawsky, S. Lin, B. Zhu, C. Chang, T. Zhou, L. Zhang and Y. He, Mol. Microbiol., 2013, 87, 80–93 CrossRef CAS.
- Y. He, J. Wu, L. Zhou, F. Yang, Y. He, B. Jiang, L. Bai, Y. Xu, Z. Deng, J. Tang and L. Zhang, Mol. Plant-Microbe Interact., 2011, 24, 948–957 CrossRef CAS.
- T. S. Peat, S. Balotra, M. Wilding, N. G. French, L. J. Briggs, S. Panjikar, N. Cowieson, J. Newman and C. Scott, Mol. Microbiol., 2013, 88, 1149–1163 CrossRef CAS.
- F. Hubrich, S. Mordhorst and J. N. Andexer, Bioorg. Med. Chem. Lett., 2013, 23, 1477–1481 CrossRef CAS.
- L. Dong and Y. Liu, Proteins: Struct., Funct., Bioinf., 2017, 85, 1146–1158 CrossRef CAS.
- M. A. Gregory, H. Petkovic, R. E. Lill, S. J. Moss, B. Wilkinson, S. Gaisser, P. F. Leadlay and R. M. Sheridan, Angew. Chem., Int. Ed., 2005, 44, 4757–4760 CrossRef CAS.
- L. C. Blasiak and J. Clardy, J. Am. Chem. Soc., 2010, 132, 926–927 CrossRef CAS.
- W. Zhang, B. D. Ames and C. T. Walsh, Biochemistry, 2011, 50, 5401–5403 CrossRef CAS.
- J. Zaitseva, J. Lu, K. L. Olechoski and A. L. Lamb, J. Biol. Chem., 2006, 281, 33441–33449 CrossRef CAS.
- J. Olucha, A. N. Ouellette, Q. Luo and A. L. Lamb, Biochemistry, 2011, 50, 7198–7207 CrossRef CAS.
- J. L. Pawlak, R. E. Padykula, J. D. Kronis, R. A. Aleksejczyk and G. A. Berchtold, J. Am. Chem. Soc., 1989, 111, 3374–3381 CrossRef CAS.
- W. J. Guilford, S. D. Copley and J. R. Knowles, J. Am. Chem. Soc., 1987, 109, 5013–5019 CrossRef CAS.
- B. Ganem and G. W. Holbert, Bioorg. Chem., 1977, 6, 393–396 CrossRef CAS.
- G. W. Holbert, L. B. Weiss and B. Ganem, Tetrahedron Lett., 1976, 17, 4435–4438 CrossRef.
- W. Jiao, E. J. Lang, Y. Bai, Y. Fan and E. J. Parker, Curr. Opin. Struct. Biol., 2020, 65, 159–167 CrossRef CAS.
- K. Helmstaedt, S. Krappmann and G. H. Braus, Microbiol. Mol. Biol. Rev., 2001, 65, 404–421 CrossRef CAS.
- S. Munack, K. Roderer, M. Ökvist, J. Kamarauskaite, S. Sasso, A. van Eerde, P. Kast and U. Krengel, J. Mol. Biol., 2016, 428, 1237–1255 CrossRef CAS.
- N. J. Blackmore, A. R. Nazmi, R. D. Hutton, M. N. Webby, E. N. Baker, G. B. Jameson and E. J. Parker, J. Biol. Chem., 2015, 290, 18187–18198 CrossRef CAS.
- F. Semmelmann, K. Straub, J. Nazet, C. Rajendran, R. Merkl and R. Sterner, J. Mol. Biol., 2019, 431, 2718–2728 CrossRef CAS.
- A. Srivastava and S. Sinha, Mol. BioSyst., 2017, 13, 142–155 RSC.
- G. Bashiri, J. M. Johnston, G. L. Evans, E. M. M. Bulloch, D. C. Goldstone, E. N. M. Jirgis, S. Kleinboelting, A. Castell, R. J. Ramsay, A. Manos-Turvey, R. J. Payne, J. S. Lott and E. N. Baker, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2015, 71, 2297–2308 CrossRef CAS.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS.
- N. G. Schmidt, E. Eger and W. Kroutil, ACS Catal., 2016, 6, 4286–4311 CrossRef CAS.
- J. Tröndle, K. Schoppel, A. Bleidt, N. Trachtmann, G. A. Sprenger and D. Weuster-Botz, J. Biotechnol., 2020, 307, 15–28 CrossRef.
- M. Jiang and H. Zhang, Curr. Opin. Biotechnol., 2016, 42, 1–6 CrossRef CAS.
- A. Kurutsch, M. Richter, V. Brecht, G. A. Sprenger and M. Müller, J. Mol. Catal. B: Enzym., 2009, 61, 56–66 CrossRef CAS.
- M. Schapfl, S. Baier, A. Fries, S. Ferlaino, S. Waltzer, M. Müller and G. A. Sprenger, Appl. Microbiol. Biotechnol., 2018, 102, 8359–8372 CrossRef CAS.
- A. Dübler, P. Krastel, H. G. Floss and A. Zeeck, Eur. J. Org. Chem., 2002, 983–987 CrossRef.
- A. W. Puri, E. Mevers, T. R. Ramadhar, D. Petras, D. Liu, J. Piel, P. C. Dorrestein, E. P. Greenberg, M. E. Lidstrom and J. Clardy, J. Am. Chem. Soc., 2018, 140, 2002–2006 CrossRef CAS.
- S. H. Akone, A. Mándi, T. Kurtán, R. Hartmann, W. Lin, G. Daletos and P. Proksch, Tetrahedron, 2016, 72, 6340–6347 CrossRef CAS.
- E. Ancheeva, L. Küppers, S. H. Akone, W. Ebrahim, Z. Liu, A. Mándi, T. Kurtán, W. Lin, R. Orfali, N. Rehberg, R. Kalscheuer, G. Daletos and P. Proksch, Eur. J. Org. Chem., 2017, 3256–3264 CrossRef CAS.
Footnotes |
| † Dedicated to Professors Heinz G. Floss and Eckhard Leistner for their inspiring research into chorismate-converting enzymes. |
| ‡ In other publications, para-aminobenzoate synthase is counted towards chorismate-converting enzymes, although these enzymes reactions do not act directly from chorismate (1) and isochorismate (2). |
| § From here on named chorismate-converting enzymes to simplify reading and understanding of the text. |
| ¶ Anthranilate synthases: Gln236/Thr425/His398 in TrpE-I, isochorismate-pyruvate lyases: Gln90 in PchB, salicylate synthases: Lys205/Thr361/Lys438 in MbtI. |
| This journal is © The Royal Society of Chemistry 2021 |