
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA–polymer conjugates via the graft-through polymerisation of native DNA in water†
Lucy A.
Arkinstall
,
Jonathan T.
Husband
 ,
Thomas R.
Wilks
,
Jeffrey C.
Foster
* and
Rachel K.
O'Reilly
*
,
Thomas R.
Wilks
,
Jeffrey C.
Foster
* and
Rachel K.
O'Reilly
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: r.oreilly@bham.ac.uk; j.c.foster@bham.ac.uk
First published on 24th April 2021
Abstract
The direct, graft-through, ring-opening metathesis polymerisation (ROMP) of unprotected DNA macromonomers is reported. By tuning the polymerisation conditions, good control is achieved, enabling the rapid and efficient synthesis of DNA-containing bottlebrush copolymers, without the need for protection of the DNA bases.
Nucleic acids underpin many of the most promising emerging technologies of the 21st century. The highly specific and programmable nature of base pairing has led to their use in a wide variety of fields, from therapeutics1 to structural nanotechnology.2 For many of these applications, it is desirable to conjugate nucleic acids to other chemical entities, to impart additional functionality or improve stability.3 In particular, attachment of synthetic polymers has emerged as a versatile way of achieving both of these goals.4–6 For example, poly(ethylene glycol) (PEG) has been used to improve nucleic acid serum stability,7–11 and thermoresponsive polymers such as poly(N-isopropylacrylamide) have been used to develop simple precipitation-based assays.12–15 The self-assembly properties of synthetic polymers have also been exploited to create nucleic acid nanoparticles that change shape in response to various biological stimuli,16,17 act as scaffolds for chemical reactions,18 and confer resistance to degradation and improved drug delivery efficiency.7,8,11
The majority of nucleic acid–polymer conjugates are linear in nature, with a single polymer attached to one end (or occasionally in the middle) of a single strand of nucleic acid. By contrast, a much wider range of architectures have been explored for synthetic polymers, ranging from stars to hyperbranched and dendritic structures, and these different shapes have been found to impart different properties.19–22 In a particularly relevant recent example, Pearce et al. demonstrated how polymer architecture has a profound effect on biological processing.23 It is reasonable, therefore, to suppose that moving away from linear nucleic acid–polymer conjugates to more complex architectures might facilitate access to novel and unique properties. We were particularly interested in the synthesis of bottlebrush-type polymers, since the brush architecture can surround the oligonucleotide with a higher local density of polymer chains, preventing protein access whilst still allowing DNA hybridisation.24–27
There are a handful of examples of DNA-containing bottlebrush polymers (DNA-BBPs) in the literature, which have been synthesised via three main routes. In the ‘grafting to’ approach, DNA is coupled to a pre-made BBP. For example, Zhang and coworkers prepared DNA-brush polymers by conjugating amino-DNA to NHS esters along the polymer backbone.24 However, coupling efficiencies are often poor for DNA–polymer coupling in solution.28 The ‘grafting from’ approach overcomes this limitation by forming the polymer in situ, using an appropriately functionalised DNA strand as a macroinitiator for a controlled polymerisation. For example, linear DNA-polymer conjugates have been prepared using reversible addition–fragmentation chain transfer (RAFT)29 polymerisation or atom-transfer radical polymerisation (ATRP).30 However, in these examples each DNA strand bears a single, terminal initiating group so it is confined to one end of the resulting BBP only. The third approach, ‘grafting through’, allows incorporation of the DNA strand anywhere within the bottlebrush structure. The groups of Herrmann and Zhang independently demonstrated the graft through ROMP of norbornene-functionalised DNA to prepare DNA-brush polymers.31,32 Grafting through makes use of DNA macromonomers (i.e. DNA strands terminated with a polymerisable unit), and can achieve much higher incorporation efficiency than either grafting to or grafting from. However, all examples of this strategy rely on the polymerisation of protected DNA, which necessitates time-consuming and yield-limiting additional steps to produce the native DNA-BBP. We therefore set out to develop a method for the graft through polymerisation of native DNA, and here report the direct production of DNA-BBPs in a single step.
We recently reported a straightforward method to conduct ROMP in water, which exploits a macroinitiator approach: a water-soluble BBP is first synthesised in organic solvent, and then transferred into aqueous buffer for chain extension (Fig. 1).33 Our previous work has demonstrated the efficient polymerisation of norbornene-terminated PEG to produce BBPs in water.34
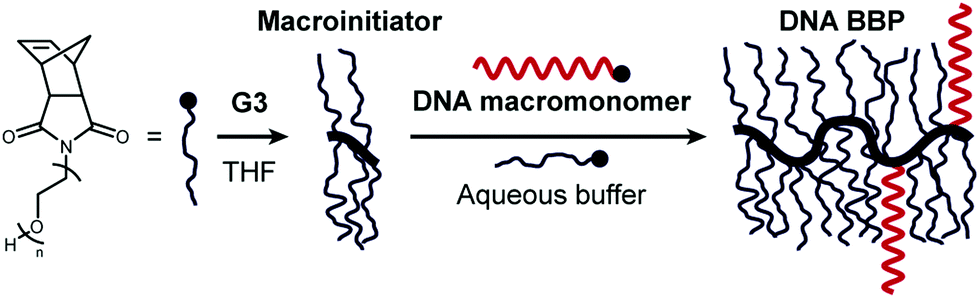 | ||
| Fig. 1 Synthesis of DNA-containing bottlebrush polymers via a macroinitiator approach in water. A water-soluble macroinitiator is first synthesised by ROMP using Grubbs’ third generation catalyst (G3), and then transferred to an aqueous solution for chain extension. Use of a norbornene-terminated DNA strand allows direct access to the desired DNA-polymer conjugates without the requirement for protection. | ||
We reasoned that the direct polymerisation of appropriately functionalised DNA could be possible using the same approach (Fig. 1). A DNA macromonomer was synthesised by coupling carboxy-functionalised exo-norbornene to a commercially available amino-modified DNA strand tagged with a fluorescent tetramethylrhodamine (TAMRA) group (*DNA-NH2) via a 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC)-mediated amidation.28 The desired DNA macromonomer (*DNA-Nb) was obtained in near quantitative conversion and utilised without further purification.
Polymerisation of DNA-Nb using the macroinitiator strategy was next attempted. The macroinitiator was synthesised by polymerisation of norbornene-functionalised PEG (PEG-Nb) using Grubbs’ third generation catalyst (G3) in tetrahydrofuran (THF).33 We used two different molecular weights of PEG–350 Da (PEG[350]-Nb) and 2000 Da (PEG[2000]-Nb)–to allow the effects of side-chain length on the ROMP process and DNA-BBP behaviour to be investigated.
Before continuing to polymerise the DNA macromonomer, we investigated whether the presence of the DNA bases would disrupt ROMP. We performed chain extension of the macroinitiator with PEG[350]-Nb in water in the presence of the four nucleobases adenine, guanine, cytosine and thymine, and examined the resulting BBP via size exclusion chromatography (SEC) in N,N-dimethylformamide (DMF) (Fig. S11a, ESI†). The presence of 100 nmol of the four nucleobases led to a loss of control over the polymerisation, evidenced by a much broader molecular weight distribution and low conversions compared to the polymerisation conducted in their absence (Table S1, ESI†). The polymerisation was repeated in the presence of the individual nucleobases (Fig. S11b, ESI†) and this revealed that guanine was responsible for the loss of control. We hypothesised that this resulted from chelation of the catalyst by the pyrimidine.35 We attempted to block this chelation by the addition of succinimide, which has previously been employed by Sleiman and coworkers for a similar purpose.35 As anticipated, a small amount (0.25 equiv. relative to *DNA-Nb) of succinimide restored quantitative monomer conversion, although the molecular weight distribution remained broad (Fig. 2).
 | ||
| Fig. 2 Effect of the presence of the nucleobase guanine (G) on macroinitiator chain extension, measured by SEC. A clear loss of control was observed when guanine was present (blue trace). Recovery of polymerisation control was possible by addition of succinimide (brown trace). Eluent: DMF + 5 mM NH4BF4, PMMA standards. | ||
We next investigated the stability of the DNA macromonomer under the polymerisation conditions. The chain extension step required addition of a THF solution of the macroinitiator to aqueous phosphate buffer, acidified to pH 2 with HCl. The low pH and presence of chloride ions are essential to allow productive metathesis to occur,36 but we were concerned that they could also cause DNA degradation. The precursor *DNA-NH2 (lacking the norbornene group) was incubated under the ROMP chain extension conditions for 1 h in the presence of the macroinitiator poly(PEG[350]-Nb)10. Liquid chromatography–mass spectrometry (LC–MS) showed no change in the main DNA peak (Fig. S12a, ESI†). While several new peaks were observed at later retention times, these were attributed to the macroinitiator rather than to DNA degradation products, which were expected to appear at earlier retention times. The mixtures were also examined by denaturing polyacrylamide gel electrophoresis (PAGE), which also showed no significant changes to the DNA (Fig. S12b, ESI†). We therefore concluded that the DNA remained stable under the ROMP chain extension conditions.
Having optimised the ROMP conditions and confirmed DNA stability, chain extension of the macroinitiators was attempted. Due to the small amount of DNA-Nb available, we chose to conduct copolymerisations with PEG-Nb and to use a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 feed of macroinitiator to DNA-Nb, to allow the polymerisations to be conducted at an accessible scale. Two DNA-PEG BBPs were targeted: poly(PEG[350]-Nb)10-b-poly(*DNA-Nb-co-PEG[350]-Nb)100 and poly(PEG[2000]-Nb)5-b-poly(*DNA-Nb-co-PEG[2000]-Nb)25 (DNA-BBP1 and DNA-BBP2, respectively). Two control BBPs lacking DNA were also prepared for comparison: poly(PEG[350]-Nb)10-b-poly(PEG[350]-Nb)100 and poly(PEG[2000]-Nb)5-b-poly(PEG[2000]-Nb)25 (BBP1 and BBP2, respectively). For all polymers, the macroinitiator was added to pH2 phosphate buffer, where the chain extension was performed with the appropriate monomer(s). Lower degrees of polymerisation (DPs) were targeted when using PEG[2000]-Nb because its large steric bulk prevents quantitative monomer conversion at higher DPs.
:1 feed of macroinitiator to DNA-Nb, to allow the polymerisations to be conducted at an accessible scale. Two DNA-PEG BBPs were targeted: poly(PEG[350]-Nb)10-b-poly(*DNA-Nb-co-PEG[350]-Nb)100 and poly(PEG[2000]-Nb)5-b-poly(*DNA-Nb-co-PEG[2000]-Nb)25 (DNA-BBP1 and DNA-BBP2, respectively). Two control BBPs lacking DNA were also prepared for comparison: poly(PEG[350]-Nb)10-b-poly(PEG[350]-Nb)100 and poly(PEG[2000]-Nb)5-b-poly(PEG[2000]-Nb)25 (BBP1 and BBP2, respectively). For all polymers, the macroinitiator was added to pH2 phosphate buffer, where the chain extension was performed with the appropriate monomer(s). Lower degrees of polymerisation (DPs) were targeted when using PEG[2000]-Nb because its large steric bulk prevents quantitative monomer conversion at higher DPs.
Based on the macroinitiator:DNA-Nb feed ratio, we expected approximately 10% of the polymer chains in each DNA-BBP sample to contain a DNA strand. We looked for evidence of these DNA-containing species by size exclusion chromatography (SEC) in DMF, using both refractive index (RI) detection and UV-visible light absorbance. A new, weak high molecular weight peak was observed in the RI traces of DNA-BBP1 and DNA-BBP2 which was absent in the control polymers, BBP1 and BBP2 (Fig. S15 and S16, ESI†). The new peaks also exhibited strong absorbance at 559 nm (Fig. 3), the absorbance maximum of the TAMRA group attached to the DNA macromonomer. We therefore concluded that the new peaks were associated with the successfully synthesised DNA-BBPs. The large change in retention volume relative to the polymer lacking DNA was unexpected but could be attributed to DNA aggregation under the SEC conditions.
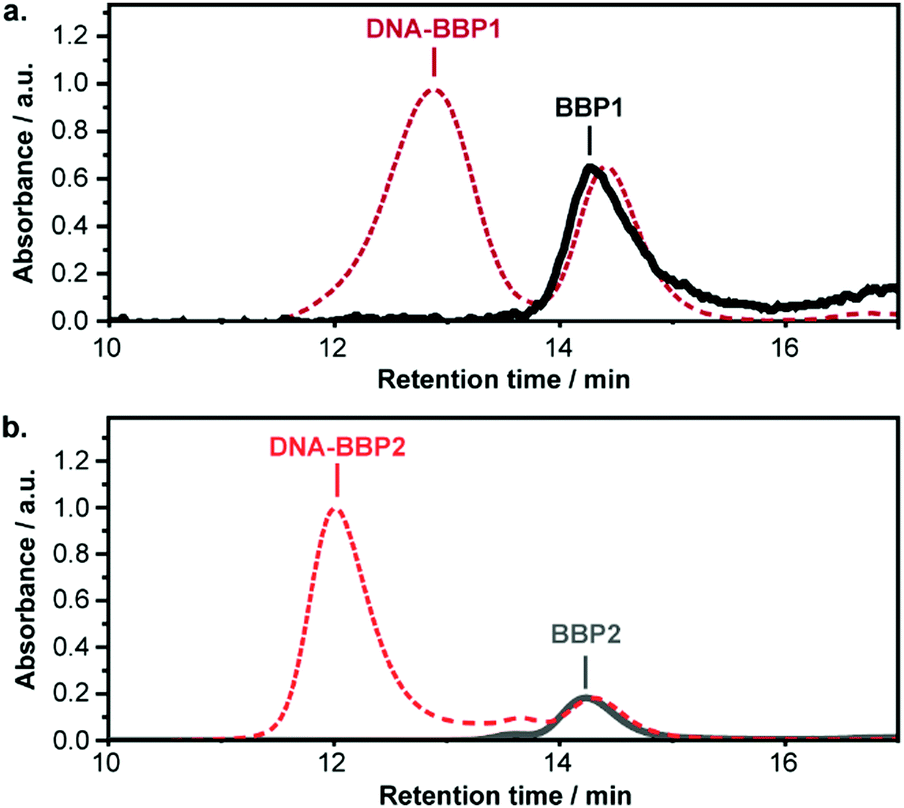 | ||
| Fig. 3 DMF SEC analysis of DNA-BBPs with UV-vis detection at 559 nm. Each DNA-BBP (dash line) is compared with the analogous DNA-free control polymer (solid line). (a) DNA-BBP1 and BBP1. (b) DNA-BBP2 and BBP2. All plots have been normalised to the BBP peak. | ||
The polymers were also investigated by denaturing PAGE (Fig. 4). Initial investigations revealed that TAMRA fluorescence was quenched in the presence of the Ru catalyst (Fig. S17, ESI†). It was therefore necessary to use high loading concentrations, leading to skewed bands on the resulting gels. Nevertheless, a new, low mobility band was clearly observed for DNA-BBP1 and DNA-BBP2 (Fig. 4a and b, lane 5). Free DNA was observed in both cases, and was removed via prep-SEC (Fig. S18, ESI†). Densitometry was performed (Fig. 4) but the aforementioned quenching effect in the DNA-BBPs meant that this most likely resulted in a large over-estimate of the amount of free DNA. The new low mobility band was not observed for the control polymers lacking DNA (lane 2), nor when the control polymers were mixed with *DNA-NH2 after polymerisation (lane 3). We therefore concluded that this band was due to the DNA-containing BBPs, which migrated slowly due to their high molecular weight. Taken together with the SEC data above, we concluded that the graft through ROMP of native DNA in water had been successful.
 | ||
| Fig. 4 Denaturing PAGE analysis of (a) DNA-BBP1 and (b) DNA-BBP2 visualised by TAMRA fluorescence. In both cases the lanes are as follows: 1 = *DNA-NH2; 2 = DNA-free BBP (BBP1 or BBP2 as appropriate); 3 = *DNA-NH2 + DNA-free BBP mixed after polymerisation; 4 = *DNA-NH2 + DNA-free BBP mixed during polymerisation; 5 = DNA-BBP (DNA-BBP1 or DNA-BBP2). Densitometric plots are included to the right of each gel, with the following band densities recorded: (a) 41% DNA-BBP1, 59% free DNA. (b) 15% DNA-BBP2, 85% free DNA. | ||
As an additional control during PAGE analysis, we mixed *DNA-NH2 into the reaction mixture during ROMP of the appropriate PEG-Nb. As expected, for PEG[2000]-Nb no additional bands were observed (Fig. 4b, lane 4). However, to our surprise for PEG[350]-Nb a faint, low-mobility band appeared (Fig. 4a, lane 4). Since the introduced DNA was incapable of covalent attachment to the polymer via ROMP, we hypothesised that this band arose as a result of physical entrapment of DNA during the polymerisation. BBPs have previously been shown to exhibit chain collapse behaviour37 so we reasoned that this may have been the origin of the entrapment effect. To test this hypothesis, two new DNA-BBPs (DNA-BBP1˙ and DNA-BBP2˙) were synthesised that were identical to DNA-BBP1 and DNA-BBP2 except for the incorporation of a small amount of an environment-sensitive dye (aminochloromaleimide, ACM38) by copolymerisation of a suitably functionalised norbornene monomer (see ESI†) during the chain extension step (Fig. 5).
 | ||
| Fig. 5 PEG chain collapse measured by changes in the fluorescence of an environment-sensitive ACM dye. The ACM monomer (black circles) and DNA-BBP2˙ (blue dots) show a decrease in fluorescence as water content increases, due to quenching of the ACM by the protic solvent. By contrast, DNA-BBP1˙ (red dots) recovers its fluorescence at high water contents, indicating chain collapse. | ||
ACMs exhibit high fluorescence emission in non-polar organic solvents, which is rapidly quenched in the presence of protic solvents, especially water. The intensity of fluorescence emission therefore provides a good indication of the relative hydrophobicity of the dye's environment. In our case, we expected PEG chain collapse to result in shielding of the dye from water and a consequent increase in fluorescence emission. DNA-BBP1˙ and DNA-BBP2˙ were dissolved at 0.33 mg mL−1 in mixtures of DMF and H2O with different H2O contents (20–100 v/v%). As expected, for DNA-BBP2˙ there was a steady decline in fluorescence emission as the water content increased (Fig. 5, blue), indicating no significant PEG chain collapse. For DNA-BBP1˙ there was a similar initial decrease in emission, but at around 70% water the trend reversed, with partial recovery of emission intensity by the time the water content reached 100% (Fig. 5, red). These results were consistent with collapse of the PEG side chains in water. As shown by the PAGE experiments above, this effect was strong enough to trap non-covalently bound DNA and was not fully disrupted even by high temperatures or strong denaturants.
To conclude, we report the first example of the direct graft-through polymerisation of native DNA in water via ROMP. This method requires no additional protection/deprotection steps and is rapid. Our results demonstrating the collapse of short PEG side chains in the presence of DNA highlight the unique behaviour that can be accessed using the bottlebrush architecture. If a way can be found to toggle this collapse on and off it could constitute a useful new strategy for the delivery of nucleic acid therapeutics. Future work will focus on incorporation of a greater proportion of DNA macromonomers into the bottlebrush product, with the eventual aim of producing BBPs in which every side chain is a DNA strand. This would open up exciting possibilities for the production of segmented structures with blocks of different DNA sequences, with the potential for controlled hierarchical assembly and programmed folding behaviour.
Conflicts of interest
There are no conflicts to declare.References
- T. C. Roberts, R. Langer and M. J. A. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- D. Yang, M. R. Hartman, T. L. Derrien, S. Hamada, D. An, K. G. Yancey, R. Cheng, M. Ma and D. Luo, Acc. Chem. Res., 2014, 47, 1902–1911 CrossRef CAS PubMed.
- M. Kwak and A. Herrmann, Angew. Chem., Int. Ed., 2010, 49, 8574–8587 CrossRef CAS PubMed.
- F. Jia, H. Li, R. Chen and K. Zhang, Bioconjugate Chem., 2019, 30, 1880–1888 CrossRef CAS PubMed.
- H. Sun, L. Yang, M. P. Thompson, S. Schara, W. Cao, W. Choi, Z. Hu, N. Zang, W. Tan and N. C. Gianneschi, Bioconjugate Chem., 2019, 30, 1889–1904 CrossRef CAS PubMed.
- J. H. Jeong, S. W. Kim and T. G. Park, J. Controlled Release, 2003, 93, 183–191 CrossRef CAS PubMed.
- J. H. Jeong, S. W. Kim and T. G. Park, Bioconjugate Chem., 2003, 14, 473–479 CrossRef CAS PubMed.
- M. Oishi, F. Nagatsugi, S. Sasaki, Y. Nagasaki and K. Kataoka, ChemBioChem, 2005, 6, 718–725 CrossRef CAS PubMed.
- M. Oishi, S. Sasaki, Y. Nagasaki and K. Kataoka, Biomacromolecules, 2003, 4, 1426–1432 CrossRef CAS PubMed.
- J. H. Jeong, S. H. Kim, S. W. Kim and T. G. Park, Bioconjugate Chem., 2005, 16, 1034–1037 CrossRef CAS PubMed.
- D. Umeno and M. Maeda, Anal. Sci., 1997, 13, 553–556 CrossRef CAS.
- D. Umeno and M. Maeda, Chem. Commun., 1998, 1433–1434 RSC.
- R. B. Fong, Z. Ding, C. J. Long, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 1999, 10, 720–725 CrossRef CAS PubMed.
- M. D. Costioli, I. Fisch, F. Garret-Flaudy, F. Hilbrig and R. Freitag, Biotechnol. Bioeng., 2003, 81, 535–545 CrossRef CAS PubMed.
- M. P. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076–5080 CrossRef CAS PubMed.
- Z. Zhao, L. Wang, Y. Liu, Z. Yang, Y.-M. He, Z. Li, Q.-H. Fan and D. Liu, Chem. Commun., 2012, 48, 9753–9755 RSC.
- F. E. Alemdaroglu, K. Ding, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2006, 45, 4206–4210 CrossRef CAS PubMed.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- Y. Zheng, S. Li, Z. Weng and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- E. Pedziwiatr-Werbicka, K. Milowska, V. Dzmitruk, M. Ionov, D. Shcharbin and M. Bryszewska, Eur. Polym. J., 2019, 119, 61–73 CrossRef CAS.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405–2420 RSC.
- A. K. Pearce, A. B. Anane-Adjei, R. J. Cavanagh, P. F. Monteiro, T. M. Bennett, V. Taresco, P. A. Clarke, A. A. Ritchie, M. R. Alexander, A. M. Grabowska and C. Alexander, Adv. Healthcare Mater., 2020, 9, 2000892 CrossRef CAS PubMed.
- X. Lu, T.-H. Tran, F. Jia, X. Tan, S. Davis, S. Krishnan, M. M. Amiji and K. Zhang, J. Am. Chem. Soc., 2015, 137, 12466–12469 CrossRef CAS PubMed.
- F. Jia, X. Lu, X. Tan, D. Wang, X. Cao and K. Zhang, Angew. Chem., Int. Ed., 2017, 56, 1239–1243 CrossRef CAS PubMed.
- F. Jia, X. Lu, D. Wang, X. Cao, X. Tan, H. Lu and K. Zhang, J. Am. Chem. Soc., 2017, 139, 10605–10608 CrossRef CAS PubMed.
- X. Lu, F. Jia, X. Tan, D. Wang, X. Cao, J. Zheng and K. Zhang, J. Am. Chem. Soc., 2016, 138, 9097–9100 CrossRef CAS PubMed.
- T. R. Wilks and R. K. O'Reilly, Sci. Rep., 2016, 6, 39192 CrossRef CAS PubMed.
- P. He and L. He, Biomacromolecules, 2009, 10, 1804–1809 CrossRef CAS PubMed.
- X. Pan, S. Lathwal, S. Mack, J. Yan, S. R. Das and K. Matyjaszewski, Angew. Chem., Int. Ed., 2017, 56, 2740–2743 CrossRef CAS PubMed.
- K. Liu, L. F. Zheng, Q. Liu, J. W. de Vries, J. Y. Gerasimov and A. Herrmann, J. Am. Chem. Soc., 2014, 136, 14255–14262 CrossRef CAS PubMed.
- X. Tan, H. Lu, Y. Sun, X. Chen, D. Wang, F. Jia and K. Zhang, Chemistry, 2019, 5, 1584–1596 CrossRef CAS PubMed.
- J. C. Foster, S. Varlas, L. D. Blackman, L. A. Arkinstall and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 10672–10676 CrossRef CAS PubMed.
- S. Varlas, J. C. Foster, L. A. Arkinstall, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O’Reilly, ACS Macro Lett., 2019, 8, 466–472 CrossRef CAS PubMed.
- H. S. Bazzi and H. F. Sleiman, Macromolecules, 2002, 35, 9617–9620 CrossRef CAS.
- J. C. Foster, M. C. Grocott, L. A. Arkinstall, S. Varlas, M. J. Redding, S. M. Grayson and R. K. O’Reilly, J. Am. Chem. Soc., 2020, 142, 13878–13885 CrossRef CAS PubMed.
- C. Li, N. Gunari, K. Fischer, A. Janshoff and M. Schmidt, Angew. Chem., Int. Ed., 2004, 43, 1101–1104 CrossRef CAS PubMed.
- Y. Xie, J. T. Husband, M. Torrent-Sucarrat, H. Yang, W. Liu and R. K. O’Reilly, Chem. Commun., 2018, 54, 3339–3342 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc08008j |
| This journal is © The Royal Society of Chemistry 2021 |