
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGas sorption of nano-porous supramolecules formed by multi-hydrogen bonded coordination capsules†
Makoto
Tadokoro
 *a,
Hironobu
Machida
a,
Ryuji
Toyofuku
a,
Megumi
Murakami
a,
Hajime
Kamebuchi
b,
Kyosuke
Isoda
c,
Fumiya
Kobayashi
a,
Kiyonori
Takahashi
d,
Shin'ichiro
Noro
e and
Takayoshi
Nakamura
d
*a,
Hironobu
Machida
a,
Ryuji
Toyofuku
a,
Megumi
Murakami
a,
Hajime
Kamebuchi
b,
Kyosuke
Isoda
c,
Fumiya
Kobayashi
a,
Kiyonori
Takahashi
d,
Shin'ichiro
Noro
e and
Takayoshi
Nakamura
d
aDepartment of Chemistry, Faculty of Science, Tokyo University of Science, Kagurazaka 1-3, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: tadokoro@rs.tus.ac.jp; Fax: +81 3 5261 4631; Tel: +81 3 5228 8714
bDepartment of Chemistry, College of Humanities and Sciences, Nihon University, Sakurajosui 3-25-40, Setagaya-ku, Tokyo 156-8550, Japan
cFaculty of Engineering and Design, Kagawa UniversityHealth Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 2217-20 Hayashi-cho, Takamatsu, Kagawa 761-0396, Japan
dLaboratory of Functional Nanomolecular Materials, Research Institute for Electronic Science, Hokkaido University, Kita 20 jo-Nishi 10 chome, Kita-ku, Sapporo 001-0020, Japan
eEnvironmental Adaptation Science, Graduate School of Environ. Sci., Faculty of Environ. Earth Science, Hokkaido Univesity, Kita 10 jo-Nishi5 chome, Kita-ku, Sapporo 060-0810, Japan
First published on 5th February 2021
Abstract
[{ReI(CO)3(Hbim)}3(tpta)]2 (1, Hbim− = 2,2′-biimidazolate monoanion, tpta = 2,4,6-tripyridyl-1,3,5-triazine) was prepared as a nano-space supramolecule by using a new group of H-bonded coordination capsules. The hamburger bun-shaped half unit [{ReI(CO)3(Hbim)}3(tpta)] contains six intermolecular H-bonds of Hbim− ligands with complementary dual NH⋯N types, and three [ReI(CO)3(Hbim)] are coordinated by bridging tridentate tpta. Interestingly, mechanical grinding easily would convert single crystals of 1 to an amorphous state with minor crystallinity while maintaining the nano-space pores. The ground sample can reversibly uptake and release small molecules such as CO2 and (CH2Cl)2.
Nano-porous molecules have multi-dimensional structures that contain molecule-sized open spaces, which allow them to uptake and release small guest molecules such as gases.1 These molecules are different from supramolecular cages or entanglements2 exhibiting porosity without pores, and possess completely closed internal voids separated from the external environment. Meanwhile, when the nano-porous molecule itself contains nano-sized open pores, it can uptake and release guest molecules not only in the crystal form but also in liquid or amorphous states.3,4
Plenty of studies of amorphous nanoporous solids have been reported either in the form of large molecules or polymers. However, in only single complexes or discreet molecules, there has been little study on the amorphous state of nano-porous solids for gas sorption, because the pore structure is mechanically collapsed in this state. Indeed, amorphous materials generally display lower levels of gas sorption.5 Meanwhile, their availability and scalability are promising from an industrial perspective, since these materials are more robust for mechanical interactions than crystalline materials. In particular, nano-porous molecules can be deliberately processed into either amorphous or crystalline states, and the amorphous state maintains the porosity even when the crystal is mechanically demolished. One of the first studies on amorphous nano-porous materials was carried out by Atwood and co-workers.6 The nano-porous molecule possessing a “noria” structure (i.e., waterwheel-shaped cyclic oligomer) has a concave hydrophobic internal cavity with a volume of ∼160 Å3 and an opening size of ∼5 Å. In the amorphous state, this molecule exhibited porosity and sorption of gases like N2 and CO2.
It is generally difficult to transform normal molecular crystals into an amorphous state, because the micro-crystalline solids maintain their microstructure or change to other more stable frameworks under heavy mechanical grinding. When the crystals of metal–organic frameworks (MOFs) are demolished by ball milling or mortar grinding, the resultant amorphous MOFs (aMOFs) tend to display drastically decreased sorption because the porous space for uptaking guest molecules is blocked or destroyed by amorphisation.7 However, Cooper et al. established a crystal-engineering method to create an amorphous solid that retains the porosity of the molecular crystal. In this chemical approach, the organic cages are desymmetrised by a dynamic covalent scrambling reaction.8 Because the product molecule contains porous spaces within itself, it can uptake and release guest molecules without forming any long-distance porous arrangement as a crystal.
In the current study, a novel nano-porous supramolecule with 2D open pores was prepared using multi-pointed H-bonds of multi-nuclear metal complexes. This supramolecule belongs to a new group of H-bonded coordination capsules. Solid-state crystals of this supramolecule can reversibly uptake and release guests, similar to some molecularly porous materials with multi-dimensional crystalline networks of nodes and struts such as MOFs,9 covalent organic frameworks (COFs),10 and hydrogen bonding organic frameworks (HOFs).11 MOFs and HOFs comprise only coordination bonding and H-bonding networks, respectively, whereas COFs are entirely covalent in nature. In contrast, the new materials proposed in this paper simultaneously use coordination and H-bonding to join the molecular building blocks, resulting in a supramolecule with a robust nano-porous structure.12
In the newly proposed supramolecular tecton,13 the [ReI(CO)3(Hbim)] unit contains not only a photoactive [ReI(CO)3]+ complex,14 but also complementary dual NH⋯N H-bonds of the didentate Hbim− (2,2′-biimidazolate monoanion) ligand. The [ReI(CO)3]+ complex coordinated with pyridyl derivatives is known to emit phosphorescence with a long lifetime and has been utilised as a photoexcited catalyst. On the other hand, some transition metal complexes with Hbim− can control their self-organised structure in the crystal by forming complementary intermolecular dual N–H⋯N H-bonds, depending on the number of Hbim− and the coordination structures.15 [ReI(CO)3(Hbim)] can coordinate to a monodentate pyridyl group and simultaneously form intermolecular connection by complementary dual N–H ⋯N H–bonds of Hbim− perpendicular to the metal coordination. If two [ReI(CO)3(Hbim)] units are joined by a didentate bridging ligand having two pyridyl groups, a molecular rectangle would be formed with two dual H-bonds between the two Hbim− ligands. In contrast, when a tridentate pyridyl ligand like 2,4,6-tripyridyl-1,3,5-triazine (tpta) is coordinated to three [ReI(CO)3(Hbim)], the result is a nano-porous supramolecule [{ReI(CO)3(Hbim)}3(tpta)]2 (1) that is shaped like a hamburger and has a triangular structure formed by three dual N–H⋯N H-bonds of Hbim− (Fig. 1). Compound 1 is a hexanuclear Re complex, with six [ReI(CO)3(Hbim)] units and strong connection through six NH⋯N H-bonds between two neutral trinuclear [{ReI(CO)3(Hbim)}3(tpta)] complexes. Interestingly, the crystal of 1 can easily be converted to the amorphous state by heavy grinding in a mortar, and the amorphous solid retaining minor crystallinity is also capable of taking up and releasing CO2 gases and (CH2Cl)2 solvent molecules through individual nano-porous molecules.
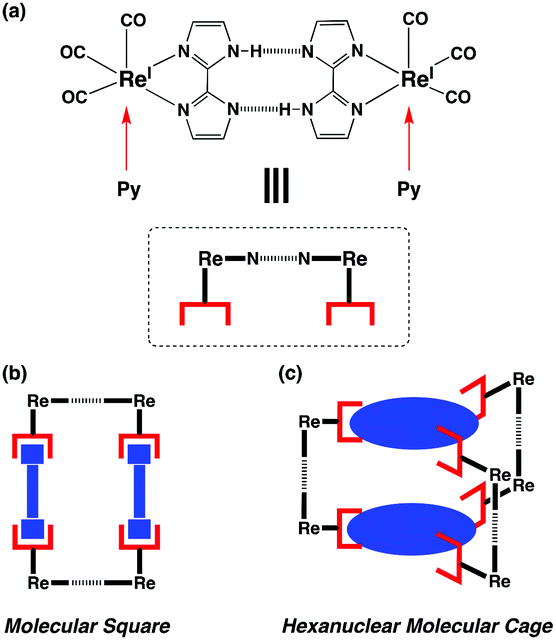 | ||
| Fig. 1 (a) The supramolecular ligating unit connected by complementary dual NH⋯N H–bonds in the [ReI(CO)3(Hbim)] complex with Hbim−. Two monodentate pyridyl groups can be individually coordinated to one unit perpendicular to the dual H-bonding plane of two Hbim− ligands. (b) The molecular square formed from two linear bidentate bridging ligands with two monodentate pyridyl groups. (c) The hexanuclear molecular cage is also constructed from two planar tridentate ligands with three pyridyl groups. | ||
Fig. 2 shows the molecular structure of 1 obtained by X-ray structure analysis. The capsule is constructed from the complementary H-bonding connections between two neutral trinuclear complexes of [{ReI(CO)3(Hbim)}3(tpta)]. First, three [ReI(CO)3(Hbim)] units are coordinated to the three pyridyl groups of tpta (cyan colour, Re(1)⋯N(5) = 2.195(10) Å). The H-bonding connections are reinforced by six complementary intermolecular NH⋯N H-bonds (red dash lines) between the three Hbim− ligands (N(2)⋯N(4)* = 2.776(19) Å) (*: −x + 4/3, −x + y + 2/3, −z + 1/6). Each ReI in 1 displays a distorted octahedral coordination consisting of three CO groups (blue sticks in Fig. 2, Re(1)⋯C(7) = 1.928(16) Å, Re(1)⋯C(8) = 1.911(16) Å, Re(1)⋯C(9) = 1.916(15) Å), a didentate Hbim− ligand (Re(1)⋯N(1) = 2.195(10) Å, Re(1)⋯N(4) = 2.168(10) Å), and a pyridyl group from tpta.
 | ||
| Fig. 2 Molecular structure of [{ReI(CO)3(Hbim)}3(tpta)]2 (1) obtained by X-ray crystal analysis. There are 18 CO groups (blue sticks), two tridentate tpta ligands (cyan), and six Hbim− groups (red), which are connected by three complementary intermolecular H–bonds of dual NH⋯N type and coordinated to 6 ReI ions. The distance of the complementary intermolecular H-bond between Hbim− ligands is N(5)⋯N(5)* = 2.776(19) Å, that between two ReI ions connected through Hbim− hydrogen bonds is Re(1)⋯Re(1)* = 10.592(3) Å (*: −x + 4/3, −x + y + 2/3, −z + 1/6), and the other distance between two ReI ions is Re(1)⋯Re(1)** = 13.332(2) Å ((**: −x + 4/3, −x + y + 2/3, −z + 1/6)). | ||
The two planar tpta ligands are arranged face-to-face, with two triazine rings slightly distorted to conform to the pore inside (distortion: 0.591(2) Å from the least-square plane based on the three pyridyl N(5) of tpta). This deviation could be caused by the six weakly enbedded (CH2Cl)2 molecules arranged in disorder around the centre of the nano-space. The electron density of the six confined (CH2Cl)2 appears fuzzy and not localised in the nano-space, and these solvent molecules do not seem to fit into periodicity of any specific space group (Fig. S1, ESI†). Another six (CH2Cl)2 were observed to localise outside the three pore openings of 1. As a result, the crystal of 1 contains layered structures with 2-D opening spaces along the ab plane (Fig. 3a).
 | ||
| Fig. 3 Two crystal structures showing stacking structure in the crystal of 1, viewed (a) along the c-axis and (b) along the b-axis from the side of the stack. Red and cyan denote different layers formed exclusively by P- and M-type isomers, respectively, and the triangle spaces in each layer contained the crystalline solvent of (CH2Cl)2. The nano-porous layer is shown by two arrow heads. | ||
Furthermore, Fig. S2 (ESI†) shows that there are two optical isomers of 1: the P-type (right-handed rotation) and M-type (left-handed rotation), which are rotational isomers when the two tpta in the same molecule are rotated against each other by 12.98(2)°. Each H-bonding (Hbim−)2 unit of 1 connected with two Hbim− ligands showed a deviation with a dihedral angle of 16.17(2)°. The crystal of 1 was constructed from 2D layers of P- and M-type isomers alternating stacked along the c axis (Fig. 3b). Each layer is constructed from 2-D hexagonal tessellation of only the P- (or M-type) isomer in the ab plane. One isomer of 1 with three triangle spaces was in contact with six of the same isomers in the layer, as shown in the view down the c-axis. Six (CH2Cl)2 molecules are contained in each triangular space, as confirmed by X-ray analysis. The (CH2Cl)2 molecules are kept in close contact by C–H⋯π and Cl⋯π interactions with the imidazolate rings of 1, respectively (C(16)⋯C(6) = 3.59(3) Å, C(17)⋯N(4) = 3.43(5) Å and C(4)⋯Cl(2) = 3.24(2) Å). Therefore, if the (CH2Cl)2 molecules are removed from the crystal of 1, extrinsic cavities are generated in the crystal (Fig. S11 and S12, ESI†). The electrospray ionisation-mass spectrum shows a peak at m/z = 3045 ([1]+), and the isotope pattern is also consistent with the peak.
Thermogravimetry (TG) measurement determined the presence of 12 (CH2Cl)2 per molecule of 1 (Fig. 4). Only half of them evaporated under atmospheric conditions at ambient temperature. Therefore, improved TG analysis was used to accurately distinguish the crystalline solvents and those easily evaporated under ambient conditions. A small amount of (CH2Cl)2 was added to an aluminium pan with crystals of 1, in order to protect the evaporation of (CH2Cl)2 in the crystal. The pan was tightly sealed with an aluminium lid. After drilling a small hole of ∼0.3 mm diameter in the lid, and the pan was maintained at 25 °C for 100 min through the TG measurement. The added (CH2Cl)2 completely evaporated to give an extra bending point on the TG/DTA trace at 15 min, six volatile (CH2Cl)2 in a periodic unit were slowly evaporated from the crystals of 1 until after 100 min. Then, the TG temperature was increased to 200 °C for 20 min, and the remaining six (CH2Cl)2 were completely evaporated. In total, a periodic unit of a crystal of 1 contains 12 (CH2Cl)2. From the X-ray crystal analysis, localised electron densities were only observed for six (CH2Cl)2, and the other six (CH2Cl)2 would exist in the nano-space of 1 in a disordered state.
 | ||
| Fig. 4 TG trace of 1. Red line: weight loss over time, blue line: temperature change. After initial evaporation of excess (CH2Cl)2 added to protect the crystals at 25 °C, the evaporation of confined (CH2Cl)2 started. Six molecules of (CH2Cl)2 in the periodic unit were lost after 100 min at room temperature, and the remaining six (CH2Cl)2 evaporated during heating at 10 °C min−1 until 200 °C. | ||
It is difficult to catch guest molecules in the interior pore of 1 by weak stacking interaction, because the H-bonding connections result in a longer bond length (Re(1)⋯Re(1)* = 10.592(3) Å (*: −x + 4/3, −x + y + 2/3, −z + 1/6)) than the coordination connections. The nano-space of 1 is too wide to connect with certain planar π-conjugated organic molecules such as pyrene and metal complexes with a square planar coordination. On the other hand, it is too narrow to include a stacking guest. Another reason why 1 fails to include some π-conjugated guests to the nano-space is the intrinsic weak interactions of H-bonds.
TG measurements were also carried out for the crystals of 1 dried in vacuo at 25 °C for 12 h. The weight loss of two (CH2Cl)2 was observed until 200 °C (Fig. S4, ESI†). From the XRD patterns, the crystal structure of 1 hardly changed after drying. The XRD patterns of the vacuum-dried crystals are slightly shifted in the wide-angle direction as a whole due to a volume contraction by solvent elimination (Fig. S5, ESI†). Accordingly, gas sorption experiments were carried out for the vacuum-dried crystals of 1 with two remaining (CH2Cl)2 in the periodic unit. The existence of two (CH2Cl)2 was also supported by adsorption and desorption results of (CH2Cl)2 in the vacuum-dried crystals of 1 at 25 °C (Fig. S6, ESI†). The saturated (CH2Cl)2 adsorption was 10 mol mol−1 (10 molecules for a periodic unit). The amount of adsorption decreases as the gas pressure of (CH2Cl)2 decreases, and it falls below 6 mol mol−1 at the end (P/P0 = 0).
Interestingly, after grinding by a mortar for ca. 40 min, crystals of 1 completely would change into an amorphous state with minor crystallinity. From the two XRD patterns in Fig. 5, the ground sample lost some reflection peaks even at a low angle, and broadened halo peaks were observed. In the SEM images, no crystalline structure could be found in the ground sample even when at the scale of 1 μm cm−1 (Fig. S7, ESI†).
 | ||
| Fig. 5 XRD patterns of (a) the amorphous solid of 1 after grinding for 30 min (blue) and (b) the crystalline solid (red). | ||
TG measurement showed no inclusion of (CH2Cl)2 in the amorphous solid of 1 (Fig. S8, ESI†), because (CH2Cl)2 in the original crystals would have evaporated when grinding destroyed the crystal structure. Elemental analysis confirmed the presence of two (CH2Cl)2 in the crystals of 1 vacuum-dried at room temperature, as well as their absence in the amorphous solid. The solid-state reflection absorption spectra of the crystalline and amorphous solids of 1 also show different patterns (Fig. S9, ESI†). Mechanical grinding with a mortar probably slightly deformed the molecular structure of 1 at the H-bonds, while a substantial part of the molecular space remained. When 1 in the amorphous state was exposed for three days to the vapour of various organic solvents (benzene, chlorobenzene, 2-PrOH, EtOH, MeOH, and (CH2Cl)2), after only (CH2Cl)2 was adsorbed, the amorphous state was changed to the crystalline form according to the XRD patterns (Fig. S10, ESI†).
Since the nano-space of 1 would not be damaged by grinding the crystals, the amorphous solid of 1 can also uptake and release CO2 molecules through the nano-spaces. Fig. 6 shows the experimental results for CO2 adsorption/desorption at −78 °C after 40 min in the crystalline and amorphous solids of 1 (both after drying in vacuo at 100 °C for 12 h). Upon initial exposure to CO2, both solids uptook CO2 rapidly, and the maximum adsorption was 5 and 3 mol mol−1 per periodic unit for the crystalline and amorphous solids of 1, respectively. Subsequently, the amount of CO2 adsorbed by both solids slowly increased. The adsorption isotherm conformed to the simple Langmuir–Blodgett curve, until the sample was filled with CO2 (12 and 10 mol mol−1 of CO2 per periodic unit for the crystals and amorphous solids, respectively). By CO2 gas, the BET surface area of crystal of 1 is 260.53 m2 g−1 and the monolayer adsorbed amount (Vm) is 49.71 cm3(STP)g−1, and the Langmuir surface area is 475.98 m2 g−1 and the monolayer adsorbed amount is 90.82 cm3(STP)g−1 (at 195 K and 100 kPa). The BET surface area of the amorphous state of 1 is 148.31 m2 g−1 and the monolayer adsorbed amount (Vm) is 28.30 cm3(STP)g−1, and the Langmuir surface area is 335.92 m2 g−1 and the monolayer adsorbed amount is 64.10 cm3(STP)g−1 (at 195 K and 101 kPa). Furthermore, both isotherms show a hysteresis during the adsorption and desorption. In the desorption process, it is also difficult to release CO2 because of their firm capture in the nano-spaces. The enhanced affinity to CO2 could be due to some chemisorption, over simple physisorption. Compared to the crystal, the amorphous solid uptakes a lower amount of CO2 at saturated adsorbed, because the random molecular arrangement in the amorphous solid structurally disturbs the movement of CO2 through it. After the CO2 adsorption experiment, the amorphous solid was partially restored to the crystalline state, since some crystalline peaks were observed in the XRD patterns. In summary, the crystal of a new nano-porous supramolecule 1 was prepared from molecular building blocks joined in HBCF (hydrogen-bonded coordination framework) by both H-bonding and coordination interactions. Such nano-space supramolecules in the amorphous state could be applied for gas sorption when they are prepared into dispersed paints or thin molecular films.
 | ||
| Fig. 6 Results of CO2 sorption by 1 at −78 °C in (a) the crystalline solid (red triangles) and (b) the ground amorphous state after 40 min (blue circles). Both samples were first dried in vacuo at 100 °C for 12 h. | ||
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. A. Little and A. I. Cooper, Adv. Funct. Mater., 2020, 1909842 CrossRef CAS.
- A. Jasat and J. C. Sherman, Chem. Rev., 1999, 99, 933 CrossRef.
- S. Jiang, K. E. Jelfs, D. Holden, T. Hasell, S. Y. Chong, M. Haranczyk, A. Trewin and A. I. Cooper, J. Am. Chem. Soc., 2013, 135, 17818 CrossRef CAS.
- N. Giri, M. G. D. Pópolo, G. Melaugh, R. L. Greenaway, K. Rätzke, T. Koschine, M. F. C. Gomes, L. Pison, A. I. Cooper and S. L. James, Nature, 2015, 527, 216–220 CrossRef CAS.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966 CrossRef CAS.
- J. Tian, P. K. Thallapally, S. J. Dalgarno, P. B. McGrail and J. L. Atwood, Angew. Chem., Int. Ed., 2009, 48, 5492 CrossRef CAS.
- T. D. Bennett and A. K. Cheetham, Acc. Chem. Res., 2014, 47, 1555 CrossRef CAS.
- S. Jiang, J. T. A. Jones, T. Hasell, C. E. Blythe, D. J. Adams, A. Trewin and A. I. Cooper, Nat. Commun., 2011, 2, 207 CrossRef.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814 CrossRef CAS.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362 RSC.
- M. Tadokoro, H. Kanno, T. Kitajima, H. S. Umemoto, N. Nakanishi, K. Isobe and K. Nakasuji, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4950 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 23116 CrossRef.
- P. N. Nguyen, H. Watanabe, Y. Tamaki, O. Ishitani and S. Kimura, Sci. Rep., 2019, 9, 11772 CrossRef.
- M. Tadokoro and K. Nakasuji, Coord. Chem. Rev., 2000, 198, 205 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, X-ray, TG, XRD, sorption, SEM, reflection spectra, ESI-mass, IR. CCDC 2047520. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc07869g |
| This journal is © The Royal Society of Chemistry 2021 |