
Regioselective side-chain amination of 2-alkyl azacycles by radical translocation: total synthesis of tetraponerine T8†
Samuel D.
Griggs
 ,
Alejandro
Martin-Roncero
,
Adam
Nelson
* and
Stephen P.
Marsden
*
,
Alejandro
Martin-Roncero
,
Adam
Nelson
* and
Stephen P.
Marsden
*
Department of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: s.p.marsden@leeds.ac.uk
First published on 18th December 2020
Abstract
The regioselective γ-C–H amination of the side-chain of saturated 2-alkyl nitrogen heterocycles is reported, proceeding through a sulfamide-directed 1,6-radical translocation. The practicality of this rapid access to 1,3-diamines is highlighted in a short synthesis of the alkaloid tetraponerine T8 and non-natural analogues.
The prevalence of nitrogen-containing molecules in nature and pharmaceuticals has inspired the development of general methods for C–N bond synthesis. A common and important structural unit found in natural products and pharmaceuticals is the 1,3-diamine motif,1 a valuable but synthetically challenging sub-class of which involves saturated nitrogen heterocycles bearing a 2-(aminoethyl) side-chain. This motif is exemplified by natural products such as cermizine D2 and the AMPA receptor antagonist kaitocephalin,3 as well as synthetic pharmaceuticals such as the antipsychotic thioridazine and the selective 5-HT7-receptor antagonist SB-269970 (Fig. 1, panel A). Simpler 2-alkyl-substituted nitrogen heterocycles are more readily synthetically accessible and occur, for example, in biologically active natural products such as coniine and solenopsin (Fig. 1, panel B). Inspired by recent advances in direct C(sp3)–H amination,4,5 we considered that the development of a method for the direct γ-amination of 2-alkyl nitrogen heterocycles would be an attractive approach to biologically-relevant 1,3-diamines (Fig. 1, panel C).
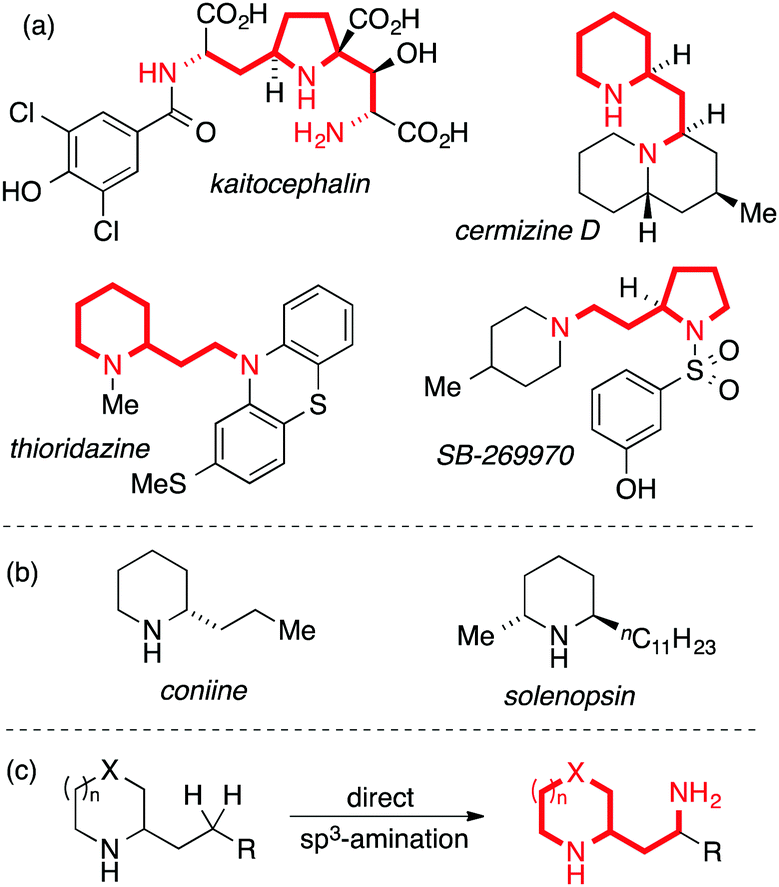 | ||
| Fig. 1 Natural products and pharmaceuticals bearing the 1,3-diamine moiety. | ||
The approach we chose to take is based upon radical translocation chemistry. The use of nitrogen-centred radicals to perform regioselective hydride abstraction is well documented: the long-established Hofmann–Löffler–Freytag reaction uses aminium radicals (photochemically-generated from N-chloroamines) to perform intramolecular 1,5-hydrogen atom transfer (HAT);6 subsequent halogen abstraction by the resulting carbon-centred radical and intramolecular substitution with the amine forms pyrrolidines. Nitrogen-centred radicals substituted with electron-withdrawing sulfonyl or carbonyl groups have subsequently been harnessed to perform a variety of synthetically useful processes initiated by 1,5-HAT through a kinetically favoured 6-membered transition state.7–13 Recently, intriguing regiocomplementary reactions have been reported, wherein N-centred radicals derived from sulfamate esters,14–22 and sulfamides22–26 have been reported instead to undergo 1,6-HAT reactions, with the change in selectivity being attributed to the long bonds and narrow bond angles in the sulfamate/sulfamide.16 The resulting radicals are frequently trapped by halogenation, with subsequent inter-24 or intramolecular14,22 C–N bond formation being observed in some cases with activated (benzylic or tertiary alkyl) substrates. While all the examples to date have utilised linear substrates, we recognised that sulfamide derivatives of cyclic amines might initiate a 1,6-HAT to alkyl substituents at the 2-position, leading potentially to side-chain functionalisation. We report herein the successful demonstration of this radical translocation method for 1,3-diamine synthesis and application in the synthesis of a member of the tetraponerine alkaloids and analogues.
At the outset, we aimed to find conditions which would lead directly to the formation of cyclic sulfamides from the parent sulfamide without isolation of N-halogenated derivatives.14,22 We therefore conducted reaction scoping on simple acyclic sulfamide derivatives. Attempts to initiate C–H amination of the phenylpropyl sulfamide 1a employing I2 and PhI(OAc)2 with heat at 50 °C resulted in a complex mixture of products. We consequently tried NaI and PhI(OAc)2, which would allow the in situ formation of I2.12 Irradiating sulfamide 1a with visible light with NaI and PhI(OAc)2 gave the C–H aminated product 2a in a 39% yield (Scheme 1). Carrying out the reaction under thermal conditions, however, gave rise to 2a in a superior 83% yield. In both cases, a single regioisomer was formed, being the expected product of 1,6-hydride abstraction.
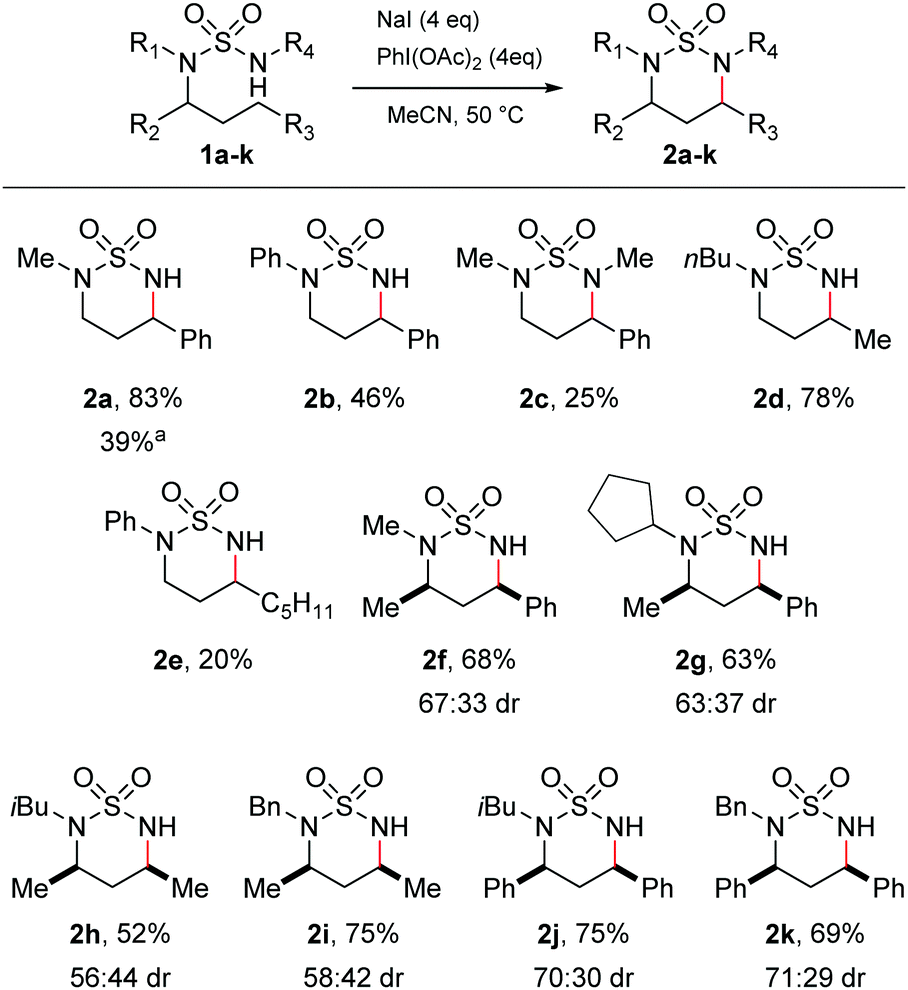 | ||
| Scheme 1 Substrate scope of acyclic sulfamides. aReaction carried out with fluorescent light instead of heating. | ||
A series of acyclic sulfamides 1b–k was then synthesised from the linear alkyl amine27,28 which varied both the non-reacting N-substituents and N-alkyl chain undergoing amination, in order to probe the substrate tolerance of the reaction. Keeping the phenylpropyl side-chain as the reactive substituent, the N-phenyl and the N,N′-dimethyl sulfamides both underwent reaction to give cyclic sulfamides 2b and 2c, albeit in lower yields than for 1a. Importantly, reaction of the N,N-dibutylsulfamide 1d gave the cyclised product 2d in a pleasing 78% yield, demonstrating that, complementary to the prior literature reports,22 cyclisation under our conditions was not limited to activated benzylic positions. A related N-phenyl sulfamide could also be cyclised (2e), but again in lower yield than for the N-alkyl derivatives.
Finally, we probed the issue of stereochemistry in the cyclisations by examining insertion to chiral variants. Cyclisation of various N,N-dialkylsulfamides bearing α-chiral substituents (2f–2k) was effected with no detriment to the reaction yields, giving rise to mixtures of diastereomeric products with moderate diastereoselectivity (up to ca. 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30). The major diastereomer was identified as the cis isomer in all cases by NMR spectroscopy (ESI†). The origins of the diastereoselectivity are uncertain. The reaction likely proceeds by initial iodination of the prochiral carbon-centred radical generated by 1,6-HAT. This process is unlikely to be stereoselective, but in the presence of excess iodide the two diastereomeric iodides may potentially be interconverted by Finkelstein substitution, with subsequent cyclisation to the diequatorial cis-disubstituted product being kinetically favoured. The higher selectivities observed for cyclisation to benzylic positions might be explained by faster interconversion of the iodides and/or an increased preference for the larger phenyl substituent (versus methyl) to be equatorial. However, since intermediates in the proposed reaction pathway were never observed, we are unable to probe these issues further.
:30). The major diastereomer was identified as the cis isomer in all cases by NMR spectroscopy (ESI†). The origins of the diastereoselectivity are uncertain. The reaction likely proceeds by initial iodination of the prochiral carbon-centred radical generated by 1,6-HAT. This process is unlikely to be stereoselective, but in the presence of excess iodide the two diastereomeric iodides may potentially be interconverted by Finkelstein substitution, with subsequent cyclisation to the diequatorial cis-disubstituted product being kinetically favoured. The higher selectivities observed for cyclisation to benzylic positions might be explained by faster interconversion of the iodides and/or an increased preference for the larger phenyl substituent (versus methyl) to be equatorial. However, since intermediates in the proposed reaction pathway were never observed, we are unable to probe these issues further.
Having successfully investigated the scope of the C–H amination in terms of substrate reactivity, attention was focused on the main goal of the research, namely to perform the side-chain amination of cyclic amines. The sulfamides of the commercially available (±)-coniine 3a and 2-(2-phenylethyl)piperidine 3b were synthesised in two steps via their sulfamoyl chlorides. Exposure of the sulfamides to the optimised reaction conditions successfully gave rise to the C–H aminated products 4a and 4b in good yields and moderate diastereoselectivity (Scheme 2). Again, complete regioselectivity was observed for the formation of the 1,6-HAT product, with no evidence of the formation of the 1,5-HAT product. X-ray crystal structures of 4a and 4b confirmed the major diastereomer to be the cis isomer (ESI†).
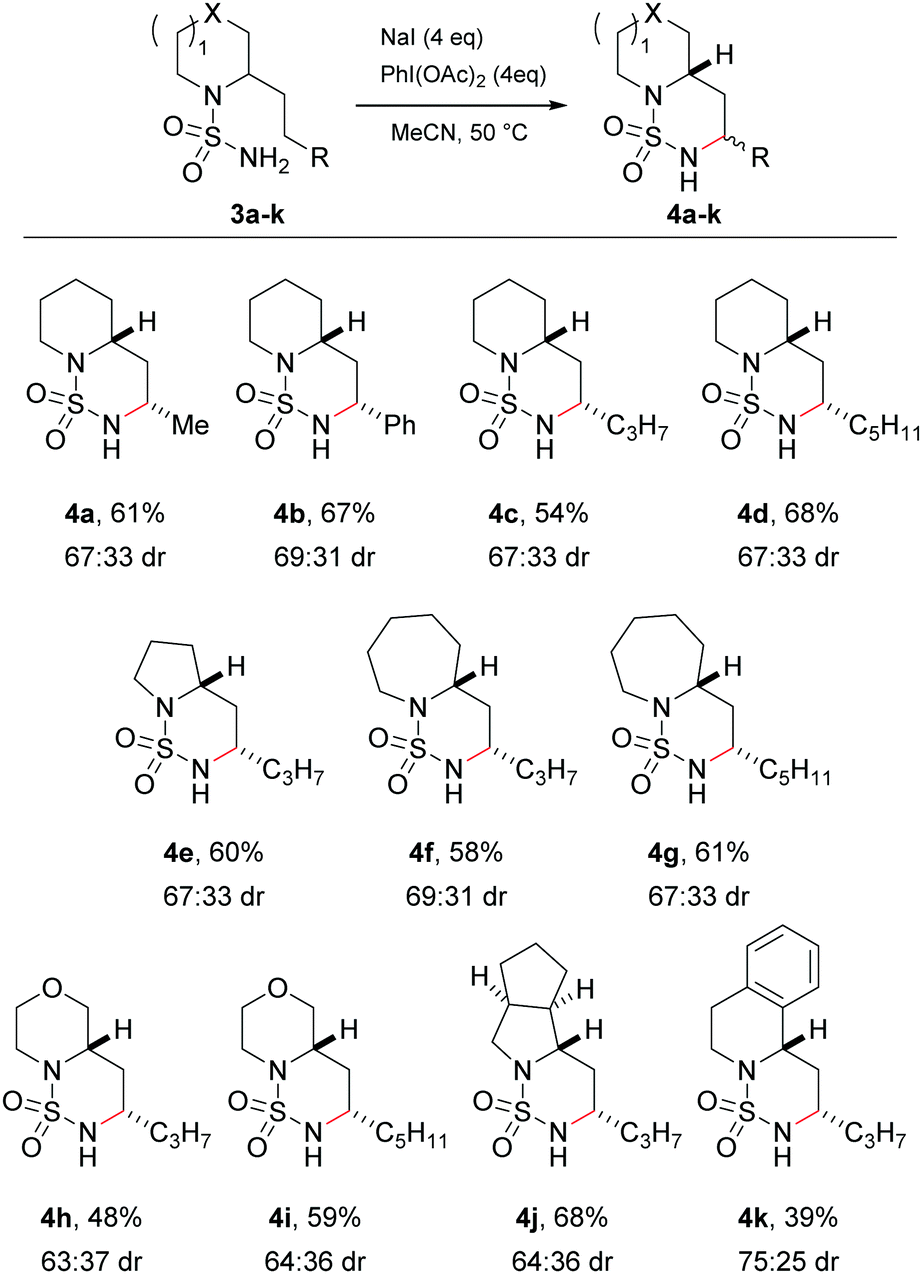 | ||
| Scheme 2 Substrate scope for the side-chain amination of 2-alkyl amines. | ||
Variation in both the side-chains and the saturated amine heterocycle was then examined more broadly. Non-commercial 2-alkyl amines were readily prepared from the simple unsubstituted cyclic amines in a single step using the method of Seidel29 prior to conversion to the sulfamide using a one-pot procedure.28 Cyclisation of the longer 2-pentyl and 2-heptyl substrates proceeded smoothly to give the C–H aminated products 4c and 4d respectively in good yields. Next, the choice of cyclic amine was investigated. The C–H amination protocol was successful with smaller (pyrrolidine, 4e) and larger (azepane, 4f and 4g) ring systems, heteroatom-containing rings (morpholine, 4h and 4i) and saturated and aromatic bicyclic structures (octahydrocyclopena[c]pyrrole, 4j, and 1,2,3,4-tetrahydroisoquinoline 4k). In all cases the major diastereomer formed in a ca. 2:1 to 3:1 ratio was the cis isomer, confirmed by X-ray analysis and/or NMR spectroscopy (ESI†). The reaction failed, however, with derivatives of N-methyl piperazine, which led to a complex mixture of products attributed to the oxidatively labile nature of the tertiary alkyl amine.
The tetraponerines 5 are a series of eight closely related tricyclic alkaloids isolated from the venom secreted by pseudomyrmecine ants of the genus Tetraponera. These compounds have been shown to display both insecticidal and cytotoxic activities, making them highly desirable targets for synthesis.30,31 There have been several reported racemic31,32 and asymmetric33–41 syntheses of the tetraponerine alkaloids and it has been identified that 1,3-diamines of type 6 are viable precursors to the natural products (Scheme 3).31,34,35
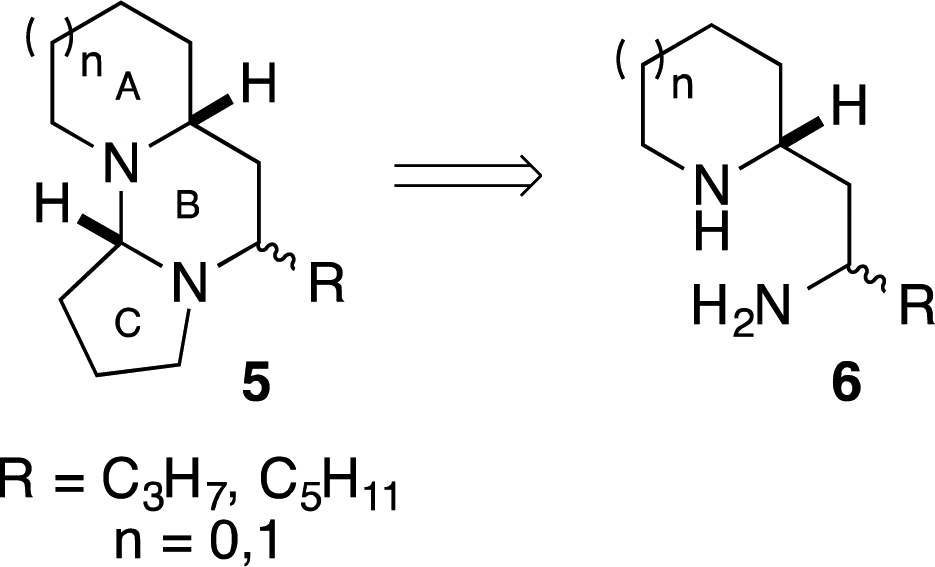 | ||
| Scheme 3 Retrosynthetic disconnection for tetraponerine alkaloid synthesis. | ||
Tetraponerine T8 5a represents the most abundant component isolated from the pseudomyrmecine venom. To demonstrate the utility of our C–H amination protocol, we sought to access 1,3-diamine 6 which would in turn allow the synthesis of the natural product. Deprotection was achieved by heating cyclic sulfamide 4d (Scheme 2) in neat 1,3-propanediamine, which resulted in transamidation to reveal the free 1,3-diamine (Scheme 4).22,24 The crude mixture was then reacted with 4-bromobutanal which initiated N-alkylation followed by subsequent aminal formation to furnish 5a, the tricyclic alkaloid (±)-T8, in just four steps from piperidine (overall 2.5% yield): to our knowledge this is the shortest synthesis of (±)-T8 from commercial materials to date.
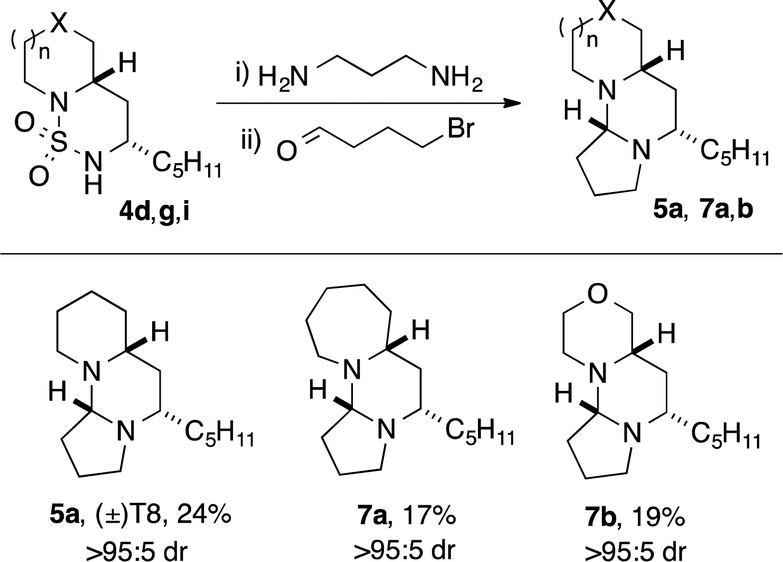 | ||
| Scheme 4 Synthesis of (±)-T8 5a and non-natural analogues. | ||
Moreover, our approach lends itself readily to the construction of non-natural analogues, of which there have been few reports in the literature.32,42 Deprotection of cyclic sulfamides 4g and 4i followed by condensation with bromobutanal gave rise to the novel analogues containing azepane 7a and morpholine 7b rings respectively, again in just four steps from the parent amine azepane/morpholine. Variation in the C-ring and B-ring substituent should be accessible employing homologous bromoalkanals43 and alternative alkyl-substituted sulfamides. This demonstrates the utility of our method for the rapid exploration of biologically-relevant chemical space.
In conclusion we have demonstrated a general and efficient method for the synthesis of 1,3-diamines by radical translocation guided by sulfamides. Most significantly, this allows for side chain functionalisation of simple 2-alkyl cyclic amines to complex 1,3-diamine derivatives. We have demonstrated the potential of this method in a concise four-step synthesis of the natural tetraponerine alkaloid T8 along with two non-natural analogues.
We thank EPSRC (EP/P016618/1 and EP/N025652/1) for funding, and Dr Chris Pask for the crystal structure determinations.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) X. Ji and H. Huang, Org. Biomol. Chem., 2016, 14, 10557–10566 RSC For examples of recent synthetic approaches, see: ; (b) C.-Y. Wu and M.-H. Xu, Org. Lett., 2019, 21, 5035–5039 CrossRef CAS; (c) K.-N. Li, A. E. Weber, L. Tseng and S. J. Malcolmson, Org. Lett., 2017, 19, 4239–4242 CrossRef CAS; (d) Y. Liu, Y.-J. Xie, H.-L. Wang and H.-M. Huang, J. Am. Chem. Soc., 2016, 138, 4314–4317 CrossRef CAS.
- H. Morita, Y. Hirasawa, T. Shinzato and J. Kobayashi, Tetrahedron, 2004, 60, 7015–7023 CrossRef CAS.
- H. Kobayashi, K. Shin-ya, K. Furihata, Y. Hayakawa and H. Seto, Tetrahedron Lett., 2001, 42, 4021–4023 CrossRef CAS.
- D. N. Zalatan and J. D. Bois, Top. Curr. Chem., 2010, 292, 347–378 CrossRef CAS.
- J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092–4106 RSC.
- (a) A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1883, 16, 558–560 CrossRef; (b) K. Löffler, Ber. Dtsch. Chem. Ges., 1910, 43, 2035 CrossRef; (c) M. E. Wolff, Chem. Rev., 1963, 63, 55–64 CrossRef CAS.
- C. G. Francisco, A. J. Herrera and E. Suárez, J. Org. Chem., 2003, 68, 1012–1017 CrossRef CAS.
- R. Fan, D. Pu, F. Wen and J. Wu, J. Org. Chem., 2007, 72, 8994–8997 CrossRef CAS.
- Q. Qin and S. Yu, Org. Lett., 2015, 17, 1894–1897 CrossRef CAS.
- N. R. Paz, D. Rodríguez-Sosa, H. Valdés, R. Marticorena, D. Melián, M. B. Copano, C. C. González and A. J. Herrera, Org. Lett., 2015, 17, 2370–2373 CrossRef CAS.
- C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287–8291 CrossRef.
- E. A. Wappes, S. C. Fosu, T. C. Chopko and D. A. Nagib, Angew. Chem., Int. Ed., 2016, 55, 9974–9978 CrossRef CAS.
- For a review, see: G. Kumar, S. Pradhan and I. Chatterjee, Chem. – Asian J., 2020, 15, 651–672 CrossRef CAS.
- D. N. Zalatan and J. D. Bois, Synlett, 2009, 143–146 CAS.
- S. M. Paradine, J. R. Griffin, J. Zhao, A. L. Petronico, S. M. Miller and M. C. White, Nat. Chem., 2015, 7, 987–994 CrossRef CAS.
- M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed., 2018, 57, 296–299 CrossRef CAS.
- S. Sathyamoorthi, S. Banerjee, J. Du Bois, N. Z. Burns and R. N. Zare, Chem. Sci., 2018, 9, 100–104 RSC.
- E. Del Castillo, M. D. Martínez, A. E. Bosnidou, T. Duhamel, C. Q. O’Broin, H. Zhang, E. C. Escudero-Adán, M. Martínez-Belmonte and K. Muñiz, Chem. – Eur. J., 2018, 24, 17225–17229 CrossRef CAS.
- S. K. Ayer and J. L. Roizen, J. Org. Chem., 2019, 84, 3508–3523 CrossRef CAS.
- W. Shu, H. Zhang and Y. Huang, Org. Lett., 2019, 21, 6107–6111 CrossRef CAS.
- Z.-Y. Ma, L.-N. Guo, Y. You, F. Yang, M. Hu and X.-H. Duan, Org. Lett., 2019, 21, 5500–5504 CrossRef CAS.
- K. Kiyokawa, S. Nakamura, K. Jou, K. Iwaida and S. Minakata, Chem. Commun., 2019, 55, 11782–11785 RSC.
- T. Kurokawa, M. Kim and J. Du Bois, Angew. Chem., Int. Ed., 2009, 48, 2777–2779 CrossRef CAS.
- T. Duhamel, M. D. Martínez, I. K. Sideri and K. Muñiz, ACS Catal., 2019, 9, 7741–7745 CrossRef CAS.
- M. A. Short, M. F. Shehata, M. A. Sanders and J. L. Roizen, Chem. Sci., 2020, 11, 217–223 RSC.
- D. Bafaluy, Z. Georgieva and K. Muñiz, Angew. Chem., Int. Ed., 2020, 59, 14241–14245 CrossRef CAS.
- For a 2-step synthesis: H. Lu, H. Jiang, L. Wojitas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192–10196 CrossRef CAS , followed by treatment with aq. NH4OH. See ESI†.
- For a one-pot synthesis: M. H. Bolli, C. Boss, C. Binkert, S. Buchmann, D. Bur, P. Hess, M. Iglarz, S. Meyer, J. Rein, M. Rey, A. Treiber, M. Clozel, W. Fischli and T. Weller, J. Med. Chem., 2012, 55, 7849–7861 CrossRef CAS.
- A. Paul and D. Seidel, J. Am. Chem. Soc., 2019, 141, 8778–8782 CrossRef CAS.
- (a) J. C. Braekman, D. Daloze, J. M. Pasteels, P. van Hecke, J. P. Declercq, V. Sinnwell and W. Francke, Z. Naturforsch., C: J. Biosci., 1987, 42, 627–630 CAS; (b) P. Merlin, J. C. Braekman, D. Daloze and J. M. Pasteels, J. Chem. Ecol., 1988, 14, 517–527 CrossRef CAS.
- I. Bosque, J. C. Gonzalez-Gomez, M. I. Loza and J. Brea, J. Org. Chem., 2014, 79, 3982–3991 CrossRef CAS.
- J. T. Kim, J. Butt and V. Gevorgyan, J. Org. Chem., 2004, 69, 5638–5645 CrossRef CAS.
- A. B. Charette, S. Mathieu and J. Martel, Org. Lett., 2005, 7, 5401–5404 CrossRef CAS.
- I. Bosque, J. C. Gonzalez-Gomez, A. Guijarro, F. Foubelo and M. Yus, J. Org. Chem., 2012, 77, 10340–10346 CrossRef CAS.
- S. G. Davies, A. M. Fletcher, I. T. T. Houlsby, P. M. Roberts and J. E. Thomson, J. Org. Chem., 2017, 82, 6689–6702 CrossRef CAS.
- R. Stragies and S. Blechert, J. Am. Chem. Soc., 2000, 122, 9584–9591 CrossRef CAS.
- J. C. O. Pacheco, A. Lipp, A. M. Nauth, F. Acke, J. P. Dietz and T. Opatz, Chem. – Eur. J., 2016, 22, 5409–5415 CrossRef.
- I. Strassnig, K. Körber, U. Hünger and H. Kunz, Synthesis, 2015, 2299–2316 CAS.
- H. Takahata, M. Kubota and N. Ikota, J. Org. Chem., 1999, 64, 8594–8601 CrossRef CAS.
- C. Yue, I. Gauthier, J. Royer and H.-P. Husson, J. Org. Chem., 1996, 61, 4949–4954 CrossRef CAS.
- P. Macours, J. C. Braekman and D. Daloze, Tetrahedron, 1995, 51(1415), 1428 Search PubMed.
- A. Rouchaud and J.-C. Braekman, Eur. J. Org. Chem., 2009, 2666–2674 CrossRef CAS.
- R. W. Alder, E. Heilbronner, E. Honegger, A. B. McEwen, R. E. Moss, E. Olefirowicz, P. A. Petillo, R. B. Sessions and G. R. Weisman, J. Am. Chem. Soc., 1993, 115, 6580–6591 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2045420–2045422. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc07625b |
| This journal is © The Royal Society of Chemistry 2021 |