
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid nanoreceptors for high sensitivity detection of small molecules by NMR chemosensing†
Federico
De Biasi‡
 *ab,
Daniele
Rosa-Gastaldo‡
*a,
Fabrizio
Mancin
a and
Federico
Rastrelli
a
*ab,
Daniele
Rosa-Gastaldo‡
*a,
Fabrizio
Mancin
a and
Federico
Rastrelli
a
aDepartment of Chemical Sciences, Università degli Studi di Padova Via Marzolo 1, 35131 Padova, Italy. E-mail: daniele.rosagastaldo@unipd.it
bDepartment of Chemistry, Università degli Studi di Firenze Via della Lastruccia 3-13, 50019 Sesto Fiorentino, Italy. E-mail: federico.debiasi@unifi.it
First published on 24th February 2021
Abstract
“Nanoparticle-assisted NMR chemosensing” combines magnetization transfer NMR techniques with the recognition abilities of gold nanoparticles (AuNPs) to isolate the NMR spectrum of relevant organic species in mixtures. The efficiency of the magnetization transfer is crucial to set the detection limit of the technique. To this aim, a second generation of nanoreceptors obtained by the self-organization of 2 nm AuNPs onto the surface of bigger silica nanoparticles shows better magnetization transfer performances, allowing the detection of analytes in water down to 10 μM concentration using standard instrumentation.
Detection and identification of selected substances in complex mixtures are primary quests in chemistry. To this aim, the most commonly used procedures rely on chromatography to break down the complexity of the problem into the analysis of many separate fractions comprising fewer components.1 However, sample manipulation in such protocols is rather time-consuming, and the possibility to directly analyse untreated samples is still highly desirable in order to optimize the experimental times and to minimize errors.
On such premises, Nuclear Magnetic Resonance (NMR) spectroscopy can be considered one of the most powerful techniques for the direct investigation of organic molecules. Nevertheless, the wealth of information it delivers when applied on plain mixtures is typically overwhelmed by spectral crowding. For this reason, many advanced NMR techniques (e.g. diffusion-ordered spectroscopy, DOSY)2,3 have been developed in the attempt to separate the spectra of the mixtures into those of the single components. In several applications the experimental output is further enhanced by the intervention of an external agent to label the signals of a target species.4–6 Nanoparticle-assisted NMR chemosensing makes no exception, as it exploits the recognition abilities of thiol-protected gold nanoparticles (AuNPs) to extract the full 1H-NMR spectrum of the target analyte that binds to the coating monolayer.5 Since the NMR signals carry unambiguous information about the interacting molecule, any interferent species, even if recognized by the nanoreceptor, can be easily identified as a false positive. Relying on a magnetization transfer driven by the nuclear Overhauser effect (NOE),7,8 this method suffers from low sensitivity, which stimulated continuous efforts over the years to decrease its limits of detection (LODs).9–11 On the NMR side, we proposed recently a new approach able to improve the detection performances down to 50 μM thanks to a high-power water-mediated Saturation Transfer Difference (HP wSTD) scheme.12 On the other hand, nanoreceptors optimization, essentially aimed at increasing the affinity for the analytes, provided so far only marginal benefits.11,13
In this communication, we present a second generation of nanoreceptors that, with no synthetic effort, optimizes the magnetization transfer efficiency in STD-based experiments, allowing us to break and further push down the LOD of nanoparticle-assisted NMR chemosensing. All the NMR experiments in this communication were performed at 25 °C on a Bruker AVANCE III spectrometer operating at 500.13 MHz 1H Larmor frequency and equipped with a 5 mm z-gradient broad-band inverse (BBI) non-cryogenic probe.
The theory of NMR relaxation predicts that the efficiency of the magnetization transfer increases as the tumbling rate of the receptor decreases. Consequently, stronger STD signals are expected upon increasing the size of the nanoreceptors (Section S1 of the ESI†).14 Nonetheless, the plain use of larger AuNPs in NMR chemosensing experiments is not trivial and brings along several drawbacks. First, the synthetic yields of large AuNPs are usually low. Second, large sizes and small coating-to-core ratios make the characterization of the monolayer much more difficult. Third, considerably higher amounts of large nanoparticles would be required since analyte binding occurs at the surface. Fourth, the affinity of the nanoreceptors for the analytes can be negatively affected by the decreased surface curvature.15
Our hypothesis was that the self-organized nanoconjugates obtained from the spontaneous assembly of a corona of small AuNPs on the surface of large inert nanoparticles could combine the benefits of both large and small nanoreceptors. To this aim, we chose two AuNPs previously used to detect designer drugs, biogenic amines and carboxylates (1-AuNPs and 2-AuNPs, Fig. 1).10,11 Since these are coated with charged thiols, they should stick to the surface of bigger inorganic nanoparticles of opposite charge. As a result, their tumbling rate in solution would be significantly lowered without affecting the local monolayer dynamics that is responsible for their binding affinity.16
 | ||
| Fig. 1 Gold nanoparticles (Section S2 of the ESI†) and analytes used in this work. 1: negatively charged AuNPs; 2: positively charged AuNPs; 3: serotonin; 4: homovanillic acid; 5: L-phenylalanine; 6: dopamine; 7: L-serine. The 1H-NMR spectrum of each analyte, complete of signals assignation, is reported in Section S6 of the ESI.† | ||
Commercially available colloidal amorphous silica nanoparticles (SiO2NPs; LUDOX® HS, negatively charged, and LUDOX® CL, positively charged) appeared to best fit our scope as they are significantly larger (about 20 nm) than the typical 2 nm AuNPs used in chemosensing applications.
As a first step, we investigated the self-organization of the AuNPs@SiO2NPs nanoconjugates. The colloidal silica suspensions in phosphate buffer were analysed using Transmission Electron Microscopy (TEM) to determine the size of the SiO2NPs. We measured an average particle diameter of 17.1 ± 2.5 nm for the LUDOX® CL and 16.9 ± 2.3 nm for the LUDOX® HS. Dynamic Light Scattering (DLS) was used to assess their hydrodynamic diameter and ζ-potential, obtaining a value of 41.1 ± 0.7 mV for the LUDOX® CL and −30.1 ± 0.8 mV for the LUDOX® HS (Section S4 of the ESI†). Upon the addition of oppositely charged AuNPs, the ζ-potentials shifted to opposite values (−22.5 ± 2.32 mV for 1-AuNPs@LUDOX® CL and 34.1 ± 1.0 mV for 2-AuNPs@LUDOX® HS), suggesting that the AuNPs were successfully assembling on the silica surface. DLS measurements revealed a slight increase of the average particles size, as detailed in Section S4 of the ESI.† Accordingly, we noticed the sedimentation of the nanoconjugates within a few minutes after the addition of the AuNPs. These effects, likely due to the cross-linking ability of the gold nanoparticles or to the overall charge neutralization of the AuNPs@SiO2NPs aggregates, were prevented by adding to the solution a little amount of polyethylene glycol (PEG2000). Besides its known ability to sterically stabilize silica colloids17 and the absence of charge (which avoids interferences in the analyte recognition process), PEG was chosen also because its 1H-NMR spectrum in water only consists in a rather sharp singlet at 3.6 ppm. DLS experiments in the presence of PEG2000 confirmed the formation of nanoconjugates with similar size and ζ-potential, without any sedimentation for at least 5 h, exceeding the time needed for sample preparation and collection of the NMR experiments (about 4 h). We further confirmed the self-organization of the AuNPs on the surface of the silica nanoparticles via TEM analysis. From the images reported in Fig. 2, it appears how the gold nanoparticles (black dots) assembled onto the surface of the silica nanoparticles (grey circles) form a scattered corona.
 | ||
| Fig. 2 TEM micrographs of the gold–silica supramolecular nanoconjugates. (a) 1-AuNPs on LUDOX® CL NPs. (b) 2-AuNPs on LUDOX® HS NPs. Gold nanoparticles appear as black spots while the larger SiO2NPs as grey circles. | ||
Another evidence of the formation of the AuNPs@SiO2NPs nanoconjugates, together with information about their composition, was obtained by 1H-NMR experiments (Section S5 of the ESI†). Small 1 μL aliquots of a 1% w/w colloid of silica nanoparticles suspended in a 0.01% w/w aqueous solution of PEG2000 were sequentially added to an NMR tube containing 500 μL of 200 μM phosphate buffer and 100 μM (in coating thiols) AuNPs in H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O = 90:10. The 1H-NMR spectra of the sample, recorded after every addition, showed a progressive disappearance of the signals of the coating monolayer due to the enhanced transverse relaxation as a result of their self-assembly onto the surface of the SiO2NPs. The NMR titrations showed also that free gold nanoparticles are not in chemical exchange with those bound to the SiO2NPs, since after each addition of the silica colloids the residual AuNPs signals exhibited only decreased intensities and not progressive broadening. Titration of 2-AuNPs with LUDOX® HS is reported in Fig. 3 as an illustrative example. In both the examined cases, completely formed nanoconjugates were characterized by an AuNPs/SiO2NPs ratio of around 33, corresponding to a footprint of about 27 nm2 per gold nanoparticle and an exclusion radius of about 3 nm, which agrees well with the AuNPs hydrodynamic radii measured via DLS (4.0 ± 1.3 nm for 1-AuNPs and 4.2 ± 0.4 nm for 2-AuNPs).
:D2O = 90:10. The 1H-NMR spectra of the sample, recorded after every addition, showed a progressive disappearance of the signals of the coating monolayer due to the enhanced transverse relaxation as a result of their self-assembly onto the surface of the SiO2NPs. The NMR titrations showed also that free gold nanoparticles are not in chemical exchange with those bound to the SiO2NPs, since after each addition of the silica colloids the residual AuNPs signals exhibited only decreased intensities and not progressive broadening. Titration of 2-AuNPs with LUDOX® HS is reported in Fig. 3 as an illustrative example. In both the examined cases, completely formed nanoconjugates were characterized by an AuNPs/SiO2NPs ratio of around 33, corresponding to a footprint of about 27 nm2 per gold nanoparticle and an exclusion radius of about 3 nm, which agrees well with the AuNPs hydrodynamic radii measured via DLS (4.0 ± 1.3 nm for 1-AuNPs and 4.2 ± 0.4 nm for 2-AuNPs).
 | ||
| Fig. 3
1H-NMR spectra of 100 μM (in coating thiols) 2-AuNPs and 200 μM phosphate buffer in H2O:D2O = 90:10 after subsequent additions of a suspension of LUDOX® HS silica nanoparticles. Asterisks denote impurities while the circle indicates the PEG2000 signal. Numbers on the right report the estimated SiO2NPs/AuNPs ratio after every addition (Section S5 of the ESI†). | ||
In the NMR titrations, sedimentation was observed after the addition of an excess of 1% w/w LUDOX® suspension, even in the presence of higher PEG2000 concentrations. This behaviour was different from that observed in the DLS experiments, where gold nanoparticles were present in slight excess after their addition and no sedimentation was observed. This suggested that stable nanoconjugates require, beside the presence of PEG, also the formation of a complete AuNPs corona on the surface of the silica nanoparticles.
Having observed the formation of stable nanoconjugates, we investigated their ability to detect selected analytes at low concentrations. 1-AuNPs@LUDOX® CL were used to analyse a mixture of 10 μM serotonin and 10 μM L-phenylalanine, 5 times lower than the previously established detection limit, by means of HP wSTD experiments.12 Results are reported in Fig. 4, along with other HP wSTD spectra acquired in the absence of nanoparticles or including either AuNPs or SiO2NPs. In agreement with theory and previous results, analyte signals were not detected in the absence of additives and in the presence of LUDOX® CL only. More remarkably, at this analyte concentration, also 1-AuNPs alone were unable to produce signals above the LOD (set as the signal intensity being larger than three times the standard deviation of the baseline noise). Only in the presence of the 1-AuNPs@LUDOX® CL nanoconjugates (panel e of Fig. 4), all the serotonin signals appeared in the final STD spectrum. Nicely enough, signals of L-phenylalanine (which has no affinity for 1-AuNPs) were not detected in any spectrum, confirming that the analyte binding selectivity of 1-AuNPs was not altered by the adsorption onto the colloidal silica for what concerns our experiments.
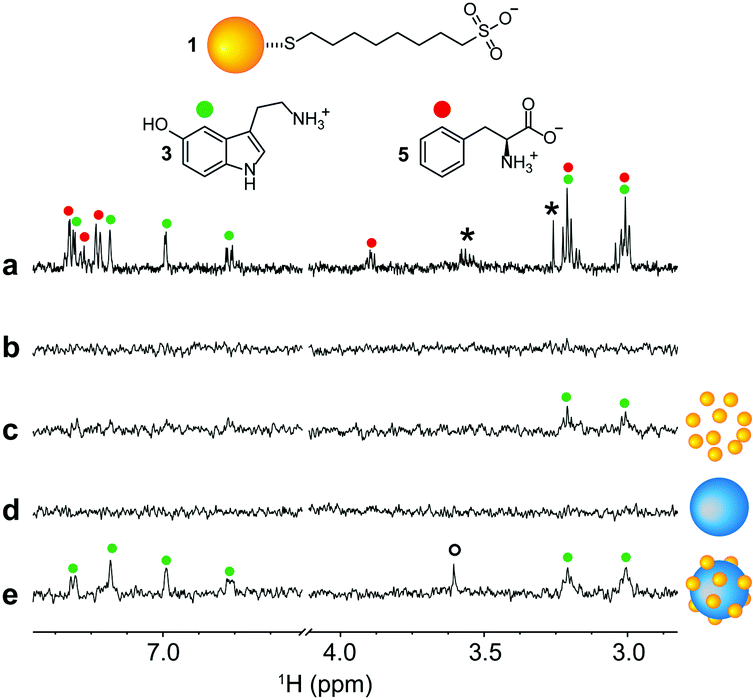 | ||
| Fig. 4 (a) 1H-NMR spectrum of 10 μM serotonin (3) and 10 μM L-phenylalanine (5) in H2O:D2O = 90:10 with phosphate buffer (200 μM, pH = 7); (b-e) HP wSTD spectra (4k scans, 4 h of acquisition time) performed on the same sample presented in (a) in the following conditions: (b) in the absence of nanoparticles; (c) in the presence of 1-AuNPs; (d) in the presence of LUDOX® CL nanoparticles; (e) in the presence of the nanoconjugates. Asterisks denote impurities while the circle indicates the resonance of PEG2000 used to stabilize the aggregates. When present, 1-AuNPs concentration was 20 μM in coating thiols. | ||
The same set of experiments was repeated using the system with opposite charges. In this case, 2-AuNPs@LUDOX® HS were used to detect 10 μM homovanillic acid, a typical catecholamine metabolite, again in the presence of 10 μM L-phenylalanine as a representative interfering species.18,19 Note that, for example, homovanillic acid concentration above 10 μM in children's urine can be related to the occurrence of neuroblastoma.20 In this case, two artefact singlets stemming from homovanillic acid do appear also in the control spectrum with no additives (panel b of Fig. 5), as well as in all the other spectra. This is because such singlets resonate too close to the water signal, and receive an extra irradiation from the fringe of the saturating field employed in HP wSTD experiments.12,21 Nonetheless, all the homovanillic acid resonances were detected only in the presence of the supramolecular nanoconjugates. Again, selectivity over non-recognized analytes was confirmed by the absence of L-phenylalanine signals in Fig. 5e. Fig. 5 also puts in evidence an additional advantage of the approach proposed here, which is the suppression of residual AuNPs signals in the HP wSTD spectra due to the enhanced transverse relaxation.
 | ||
| Fig. 5 (a) 1H-NMR spectrum of 10 μM homovanillic acid (4) and 10 μM L-phenylalanine (5) in H2O:D2O = 90:10 with phosphate buffer (200 μM, pH = 7). (b-e) HP wSTD spectra (4k scans, 4 h of acquisition time) performed on the same sample presented in (a) in the following conditions: (b) in the absence of nanoparticles; (c) in the presence of 2-AuNPs; (d) in the presence of LUDOX® HS nanoparticles; (e) in the presence of the nanoconjugates. A plus symbol is used to indicate overly enhanced resonances. Asterisks denote impurities while the circle indicates the resonance of PEG2000 used to stabilize the aggregates. When present, 2-AuNPs concentration was 20 μM in coating thiols. The broad signals in (c) and (e) indicated by the triangles stem from the monolayer of 2-AuNPs. | ||
Comforted by the retained selectivity of the AuNPs upon their interaction with the silica nanoparticles, as well as by the improved sensitivity delivered by the nanoconjugates, we repeated the HP wSTD experiments on two separate mixtures in which the targets were present at 10 μM concentration, while other three interfering molecules were present at 20 μM concentration. Both mixtures were composed by dopamine (6, target of the 1-AuNPs@LUDOX® CL nanoconjugates), homovanillic acid (4, target of the 2-AuNPs-LUDOX® HS nanoconjugates), L-phenylalanine (5) and L-serine (7). Results are presented in Fig. 6, where only the aromatic portion of the spectra is reported. Both types of nanoconjugates continued to retain their selectivity towards the respective binding candidate also in the presence of many interferent molecules at higher concentrations. Notably, Fig. 6 also demonstrates how it is possible to analyse the same sample with different nanoparticles to detect different target analytes.
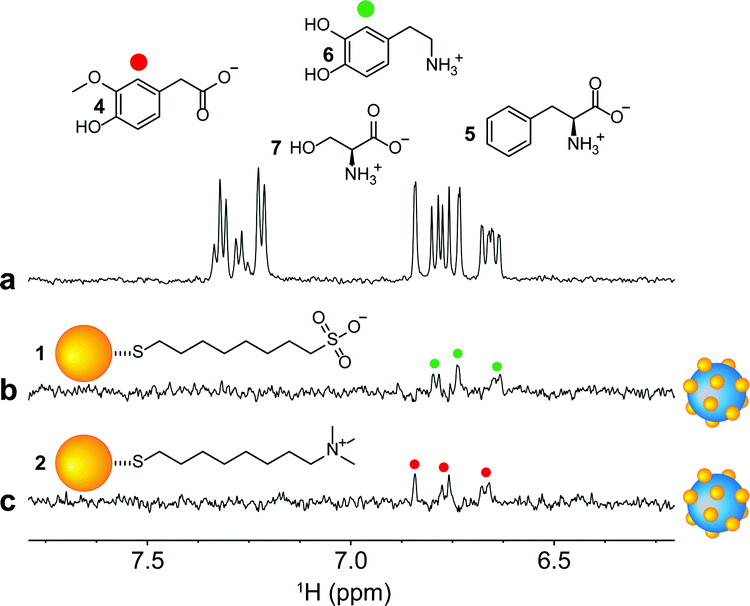 | ||
| Fig. 6 (a) 1H-NMR spectrum of a mixture of dopamine (6), homovanillic acid (4), L-phenylalanine (5) and L-serine (7) all 20 μM in H2O:D2O = 90:10 with phosphate buffer (500 μM, pH = 7); (b-c) HP wSTD spectra (4k scans, 4 h of acquisition time) performed on samples similar to that presented in (a) but in which the concentration of only the binding candidate was lowered down to 10 μM (dopamine for (b) and homovanillic acid for (c)). Both HP wSTD experiments were performed in the presence of the nanoconjugates (1-AuNPs@LUDOX® CL in (b) and 2-AuNPs@LUDOX® HS in (c)). In both HP wSTD experiments, the AuNPs concentration was 20 μM in coating thiols. A figure showing the entire NMR sweep is reported in Section S7 of the ESI.† | ||
In conclusion, we have demonstrated that self-organized nanoconjugates prepared by assembling small AuNPs on the surface of larger silica nanoparticles allowed to obtain nanoreceptors that retain the recognition properties of small monolayer protected nanoparticles while increasing their magnetization transfer ability. This approach allowed to further decrease the LOD of NMR chemosensing to 10 μM on non-cryogenic probes. The use of charged nanoparticles in the analysis of complex matrixes like urine and biofluids might not yet be straightforward, as many analytes can be detected at once because of the untargeted nature of the interaction involved in the recognition process.22,23 Nonetheless, the modularity of the proposed method allows an easy modification and fine-tuning of the sensing systems.
This work was partially supported by P-DiSC#09BIRD2017-UNIPD and P-DiSC#07BIRD2020-UNIPD granted by Università degli Studi di Padova and by PRIN 2015 RNWJAM granted by MIUR. F.M. and D.R.-G. thank the Italian Association for Cancer Research (AIRC) for financial support (IG “25003”).
Conflicts of interest
There are no conflicts to declare.Notes and references
- W. M. Niessen and A. Tinke, J. Chromatogr. A, 1995, 703, 37–57 CrossRef CAS.
- C. S. Johnson, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 203–256 CrossRef CAS.
- M. Nilsson and G. A. Morris, Chem. Commun., 2007, 933–935 RSC.
- J. Reuben, Prog. Nucl. Magn. Reson. Spectrosc., 1973, 9, 3–70 CrossRef.
- B. Perrone, S. Springhetti, F. Ramadori, F. Rastrelli and F. Mancin, J. Am. Chem. Soc., 2013, 135, 11768–11771 CrossRef CAS.
- N. K. J. Hermkens, N. Eshuis, B. J. A. Van Weerdenburg, M. C. Feiters, F. P. J. T. Rutjes, S. S. Wijmenga and M. Tessari, Anal. Chem., 2016, 88, 3406–3412 CrossRef CAS.
- A. Chen and M. J. Shapiro, J. Am. Chem. Soc., 1998, 120, 10258–10259 CrossRef CAS.
- M. Mayer and B. Meyer, Angew. Chem., Int. Ed., 1999, 38, 1784–1788 CrossRef CAS.
- M. V. Salvia, G. Salassa, F. Rastrelli and F. Mancin, J. Am. Chem. Soc., 2015, 137, 11399–11406 CrossRef CAS.
- L. Gabrielli, D. Rosa-Gastaldo, M. V. Salvia, S. Springhetti, F. Rastrelli and F. Mancin, Chem. Sci., 2018, 9, 4777–4784 RSC.
- M. V. Salvia, F. Ramadori, S. Springhetti, M. Diez-Castellnou, B. Perrone, F. Rastrelli and F. Mancin, J. Am. Chem. Soc., 2015, 137, 886–892 CrossRef CAS.
- F. De Biasi, D. Rosa-Gastaldo, X. Sun, F. Mancin and F. Rastrelli, J. Am. Chem. Soc., 2019, 141, 4870–4877 CrossRef CAS.
- X. Sun, L. Riccardi, F. De Biasi, F. Rastrelli, M. De Vivo and F. Mancin, Angew. Chem., Int. Ed., 2019, 58, 7702–7707 CrossRef CAS.
- D. Neuhaus and M. P. Williamson, The Nuclear Overhauser Effect in Structural and Conformational Analysis, Wiley-VCH, 2nd edn, 2000 Search PubMed.
- M. Lucarini, P. Franchi, G. F. Pedulli, C. Gentilini, S. Polizzi, P. Pengo, P. Scrimin and L. Pasquato, J. Am. Chem. Soc., 2005, 127, 16384–16385 CrossRef CAS.
- L. Riccardi, L. Gabrielli, X. Sun, F. De Biasi, F. Rastrelli, F. Mancin and M. De Vivo, Chem, 2017, 3, 92–109 CAS.
- E. Bourgeat-Lami, J. Nanosci. Nanotechnol., 2002, 2, 1–24 CrossRef CAS.
- S. Barco, I. Gennai, G. Reggiardo, B. Galleni, L. Barbagallo, A. Maffia, E. Viscardi, F. De Leonardis, V. Cecinati, S. Sorrentino, A. Garaventa, M. Conte and G. Cangemi, Clin. Biochem., 2014, 47, 848–852 CrossRef CAS.
- V. Strenger, R. Kerbl, H. J. Dornbusch, R. Ladenstein, P. F. Ambros, I. M. Ambros and C. Urban, Pediatr. Blood Cancer, 2007, 48, 504–509 CrossRef.
- D. F. Davidson, Ann. Clin. Biochem., 2005, 42, 200–207 CrossRef CAS.
- B. Cutting, S. V. Shelke, Z. Dragic, B. Wagner, H. Gathje, S. Keim and B. Ernst, Magn. Reson. Chem., 2007, 45, 720–724 CrossRef CAS.
- B. Zhang, M. Xie, L. Bruschweiler-Li, K. Bingol and R. Brüschweiler, Anal. Chem., 2015, 87, 7211–7217 CrossRef CAS.
- M. Xie, A. L. Hansen, J. Yuan and R. Brüschweiler, J. Phys. Chem. C, 2016, 120, 24463–24468 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc07559k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |