
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical evaluation of the carbene-based site-selectivity in gold(III)-catalyzed annulations of alkynes with anthranils†
Kaifeng
Wang
a,
Qiao
Wu
a,
Siwei
Bi
 *a,
Lingjun
Liu
a,
Guang
Chen
ab,
Yulin
Li
c,
Tony D.
James
*de and
Yuxia
Liu
*a
*a,
Lingjun
Liu
a,
Guang
Chen
ab,
Yulin
Li
c,
Tony D.
James
*de and
Yuxia
Liu
*a
aSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, 273165, P. R. China. E-mail: liuyuxia2008@163.com; siweibi@126.com; Fax: +86-537-4456305; Tel: +86-537-4458308
bMinistry of Education, Key Laboratory of Hainan Trauma and Disaster Rescue, College of Emergency and Trauma, Hainan Medical University, Haikou 571199, China
cKey Laboratory of Tibetan Medicine Research & Qinghai Key Laboratory of Qinghai-Tibet Plateau Biological Resources, Northwest Institute of Plateau Biology, Chinese Academy of Science, Xining 810001, Qinghai, P. R. China
dDepartment of Chemistry, University of Bath, Bath, UK BA2 7AY, UK
eSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, P. R. China
First published on 23rd December 2020
Abstract
The gold(III)-catalyzed annulations of alkynes with anthranils were evaluated using DFT calculations. A unified rationale for the Br-migration on α-imino gold(III)-carbene was proposed, from which an unprecedented “N-donation/abstraction substitution” mechanism was established using the substituted anthranils, while direct C–H nucleophilic attack was involved with the unsubstituted anthranils. The controlling factors guiding the site-selectivity were uncovered. These computational studies provide insight for developing new α-imino gold(III)-carbene mediated reactions.
Gold(III)-catalyzed annulations of alkynes1 with isoxazole derivatives have attracted significant interest since they provide atom- and step-economical access to diverse aza-heterocycles found in numerous natural products, organic functional materials and pharmaceuticals.2 Considerable effort has been devoted to developing gold carbene-promoted reactions3 for the selective synthesis of azaheterocyclic scaffolds. In this context, three fascinating annulations have recently been developed by the group of Hashmi to construct diverse azacycles through the tuning of nucleophilic sites for gold(III) carbenoid intermediates (Scheme 1)4,5 Using KAuBr4 as a catalyst, the reaction of substituted-anthranil 2M with N-benzyl ynamide 1a is proposed to undergo Ca-nucleophilic attack to generate the quinoline-fused polyazaheterocycle Pa (Scheme 1(1)),4 while with N-furanylmethyl ynamide 1b undergoes exclusive Cb-attack, producing 2-aminopyrrole Pb (Scheme 1(2)).4 Interestingly, the annulation of unsubstituted-anthranil 2N with ynamide 1t under AuBr3 catalysis, results in the formation of the unprotected 7-acylindole Pt, where Ct attack is favoured (Scheme 1(3)).5
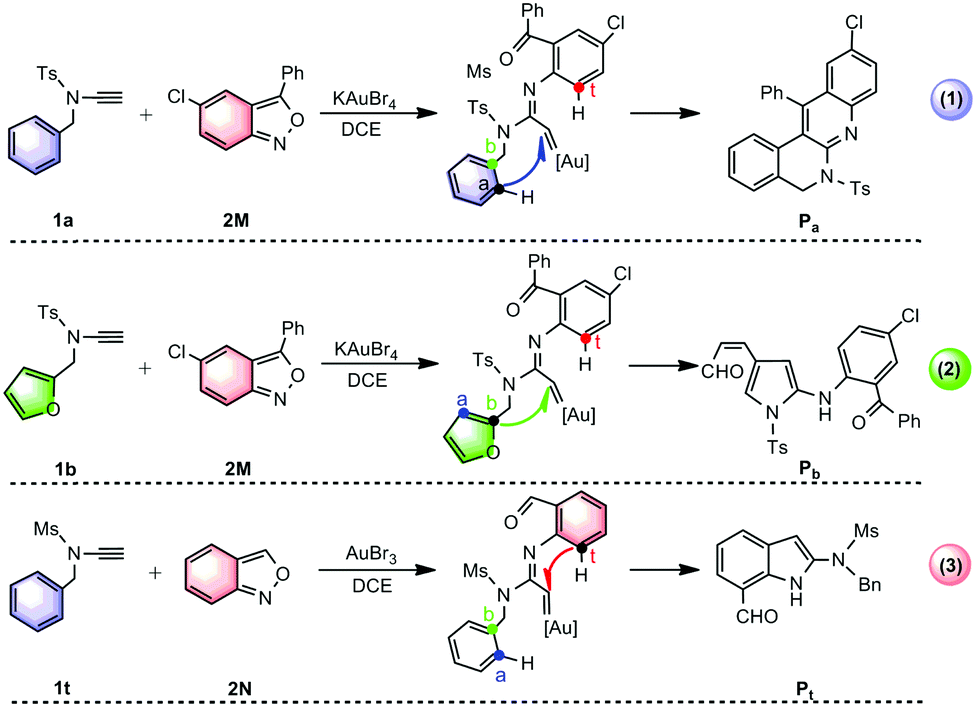 | ||
| Scheme 1 Gold(III)-catalyzed annulations of N-aryl ynamides with anthranils reported by Hashmi's group.4,5 | ||
The divergence of the nucleophilic site-selectivity at the gold carbene3 is very interesting but means that access to desired azaheterocycles in a facile and general pattern is challenging. Hence, we decided that it was important to provide an in-depth understanding of the reaction mechanisms and, especially, the inherent origins of the site-selectivity, in order to help generalize these reactions and make them predictable. Mechanistically, according to the general proposal,6 the reactions in Scheme 1 are assumed to proceed via a direct nucleophilic attack of the Ca, Cb or Ct atom at the carbene C atom. By employing an exhaustive DFT evaluation (see the Computational details in the ESI†), we explored whether the direct attack pathway or other alternative mechanisms could be used to rationalize the observed annulation products. Furthermore, the inherent controlling factors affecting the site-selectivity of these reactions were thoroughly investigated. We anticipate that these results will be informative for the future design of related catalytic reactions.
For the 1a system, the DFT-computed free energy profiles are given in Fig. 1. The reaction is initiated by coordination of KAuBr4 with the C⋯C bond of 1a to give Au⋯π coordinated complex IM1. Then the 2M N atom, acting as a nucleophile, attacks the alkyne internal C atom of IM1 due to the greater electron-deficiency of the internal C over the terminal C, as indicated by the calculated NBO charges (0.385e for the former and −0.443e for the latter). Through TS2-3, the necessary Au(III) carbenoid IM3 can then be formed by cleaving the N–O bond7,8 with a barrier of 10.9 kcal mol−1. From IM3, Ca nucleophilic attack at carbene C will exclusively produce product Pa. Therefore, we firstly evaluated the direct attack mechanism.9 Unexpectedly, the desired Ca-attack denoted as TS3-9 is 3.7 kcal mol−1 less stable than the Ct-attack viaTS3-4. Therefore, we explored a new mechanism, initiated by one Br(Au) migration. As shown in Fig. 1(black line), one Br(Au) transfers to the carbene C atom to give IM5. The calculations indicate that, through TS3-5, Br-migration occurs with a barrier of only 0.2 kcal mol−1 and is exergonic by 10.8 kcal mol−1. Subsequently, an unprecedented “SN2-type N-donation/abstraction substitution” achieves a formal Cb-attack, in which the sp2–N atom substitutes with the migrated-Br viaTS5-7 (N-donation substitution) and is then replaced by a Cb atom viaTS7-8 (N-abstraction substitution), affording the five-membered cyclized intermediate IM8. After ring expansion through the C(Au) migration to the Ca atom, the target Ca-attack complex IM9 is obtained with an energy demand of 14.6 kcal mol−1 relative to IM8. From the formal Ca-attack pathway (black line), one can clearly see that three energetically comparable TSs leading to Pa, TS3-5, TS5-7, and TS7-8, are lower in energy than TS3-4 (red line) and TS3-9 (blue line). Two crucial factors might be responsible for such energy differences. One is the extremely flat Br-migration due to much smaller structural deformation with slight change of the Br–Au–C(carbene) angle (see Fig. S1 in the ESI†). And the other may be related to the high stability of IM5 after Br-migration, which remarkably reduces the potential energy surface of subsequent processes, making the formation of Pa easier. From IM3, other possible reaction scenarios are disfavoured energetically and are given in Fig. S2 of the ESI.†
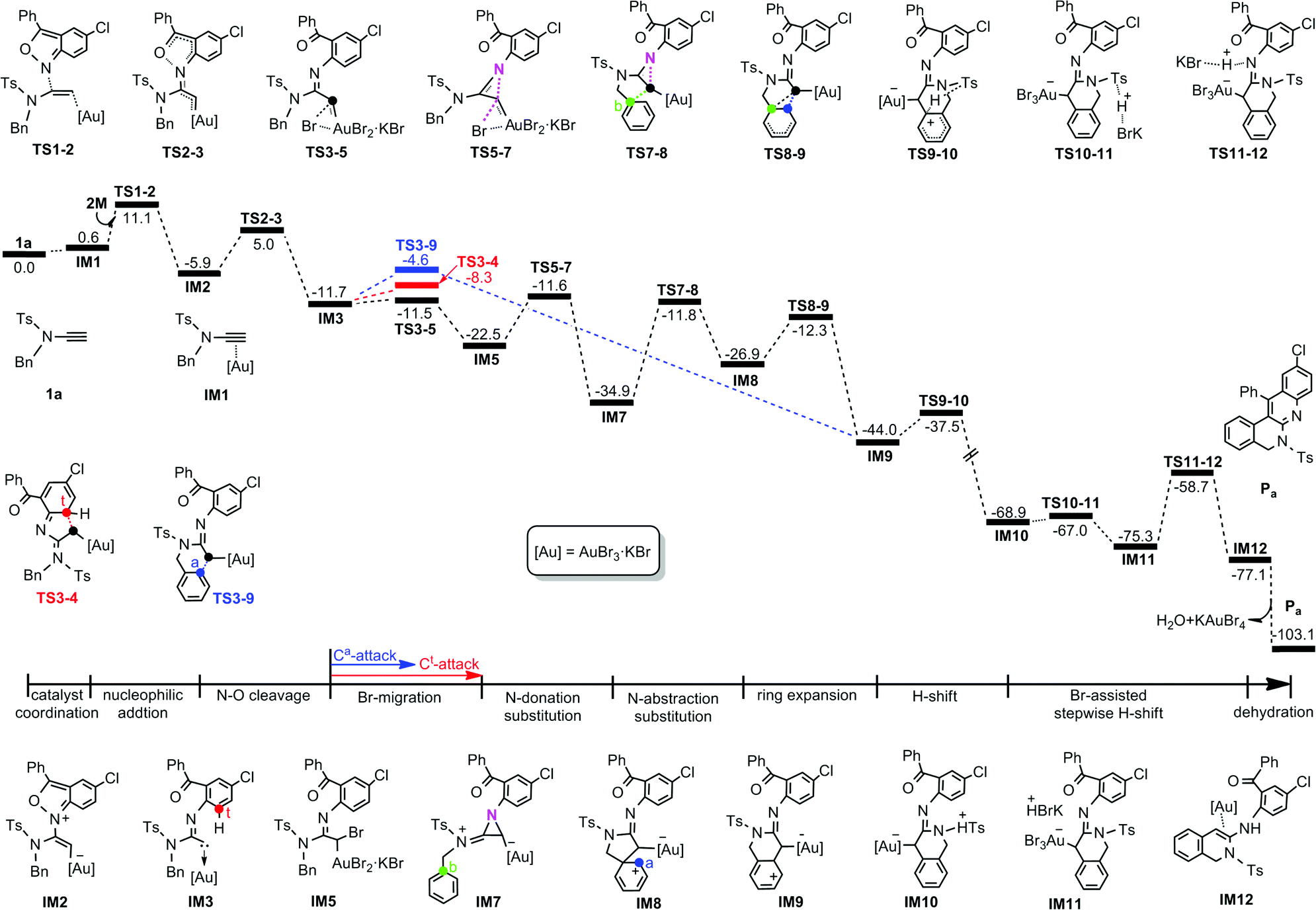 | ||
| Fig. 1 Calculated Gibbs energy profile in DCE for the formation of quinoline-fused polyazaheterocycle Pa established in the present work. The relative free energies are given in kcal mol−1. | ||
Following IM9, the Ca-attached H atom transfers to the sp2–N atom resulting in the formation of Pa. The calculated results indicate that the (Ca)H firstly transfers to one adjacent (S)O atom of the Ts fragment and then is trapped by the sp2–N atom with the assistance of KBr. The H-shift step viaTS9-10 surmounts an energy demand of 6.5 kcal mol−1 and affords the extremely exergonic intermediate IM10 owning to the formation of the aromatic benzene ring.10IM10 then experiences a KBr Br-assisted H-abstraction/donation, smoothly giving rise to the target product Pa after dehydration. Along the reaction coordinate, the Cb-substitution by the N atom, corresponding to the IM7 → IM8 transformation, is considered as the rate-determining step of the whole reaction and has a barrier of 23.1 kcal mol−1.
In the case of the 1b annulation, the proposed mechanism of the 1a-system, Au(III)-coordination → anthranil N nucleophilic addition → N–O cleavage → Br-migration → N-donation/abstraction substitution, is applied to rationalize the Cb-attack to the C(Au) atom for formation of key intermediate IM18 (1b Cycle, Fig. 2). Then, the reaction is predisposed to chemoselectively cleave the Cb–O bond of the furan moiety viaTS18-24 to provide IM26. The alternative C(Au)-migration (TS18-21, Fig. S3, ESI†) was also examined and found to be highly unfavourable (−34.7 kcal mol−1 for TS18-21 and −44.9 kcal mol−1 for TS18-24). With the Cb–O cleavage, the Br-assisted stepwise H-shift finally produces Pb and regenerates the KAuBr4 active catalyst. Finally, the 1t reaction mechanism was considered (1t Cycle, Fig. 2). After Au(III)-coordination, N nucleophilic addition, N–O cleavage and Br-migration,11 Ct-substitution with Br(Au) is selectively favoured, from which the Ct-attached H-shift and subsequent dehydration, results in Pt formation. The calculated results for other competitive pathways involved in 1b- and 1t-systems are illustrated in Fig. S4 and S5 in the ESI.†
 | ||
| Fig. 2 Plausible catalytic cycles for the Au(III)-catalyzed selective annulations of N-aryl ynamides with anthranils based on the current calculations. | ||
We then turned our attention to uncover how the nucleophilic site-selectivity at Ca, Cb and Ct atoms are controlled in these reactions. The relative free energies for the transition states (TSs) of key Br-migration, N-donation substitution and N-abstraction substitution leading to Ca/Cb selectivity, as well as that of Ct-attack resulting in Ct-selectivity are summarized in Fig. 3(i). Note that, in 1a-/1b- and 1t-systems, the N-donation substitution TSs (TS5-7/TS16-17 and TS3′-5′ in Fig. 1 and Fig. S4, S5, ESI†) exhibit very similar characteristics for the reaction regions with selected (sp2)N⋯C(Au) distances of 1.934 Å, C(Au)⋯Br distance of ∼3.000 Å and (sp2)N–C(N)–C(Au) angle of 89.6°. Intriguingly, compared with the N-donation substitution TSs, the competitive Ct-attack TSs exhibit almost opposite stability changes (0.2 vs. 3.5 in the 1a-system, −2.9 vs. 7.4 in the 1b-system and 4.9 vs. −2.7 kcal mol−1 in the 1t-system, respectively).
 | ||
| Fig. 3 (i) The relative free energies (in kcal mol−1) for the crucial site-selective transition states, as well as (ii) optimized geometries with selected structural parameters for Ct substitution transition states in 1a-/1b- and 1t-systems. The bond distance and bond angle are given in Å and °, respectively. | ||
Thus, it is predicted that the site-selectivity is closely related to the geometric constrains of the Ct-attack TS. The distinctive degree of the trans-influence of the Br atom is mainly responsible for the bifurcated selectivity at Ca/Cb and Ct atoms. It can be seen in Fig. 3 (ii) that a weaker trans-influence is exhibited in the 1t-system with a Br–C(carbene)–Ct bond angle of 151.6° (166.6° in the 1a-system and 166.4° in the 1b-system), which, in turn, makes the interaction of the Ct with the C(carbene) atom easier. As a result, smaller energy demand is required for the 1t-system. In contrast, because of the greater Br trans-influence, the resultant larger energy demand forces the 1a- and 1b-systems to follow the N-donation substitution and thus Ca/Cb selectivity is favoured.
After favourable N-donation substitution followed by N-abstraction substitution in the 1a-/1b-systems, Ca- and Cb-selectivity was further evaluated. Two selective-determining TSs for the ring-expansion with Ca-selectivity and ring-opening with Cb-selectivity, TS8-9vs.TS8-9′ in the 1a-system and TS18-21vs.TS18-24 in the 1b-system, were compared and given in Fig. 4. For the 1b-system, the main driving force for the favoured ring-opening might originate from the greater electrostatic O⋯C2 interaction.12 As shown by the calculated NBO charges, the charges on O and C2 atoms in TS18-24 are −0.45 and 0.39 e, whereas those of C(Au) and Ca in TS18-21 are −0.42 and −0.18 e, respectively. Clearly, the stronger electrostatic attraction involved in TS18-24 is more advantageous to cleave the Cb–O bond and thereby generates a relatively lower energy consumption. Nevertheless, significantly different from five-membered ring-opening in the 1b-system, TS8-9′ in the 1a-system is characteristic of a six-membered ring-opening, in which the Cb–C4 bond rupture will generate a substantially unstable terminal C cation, thereby resulting in a greater energy requirement. Consequently, the reaction is forced to follow a Ca-selective ring-expansion viaTS8-9.
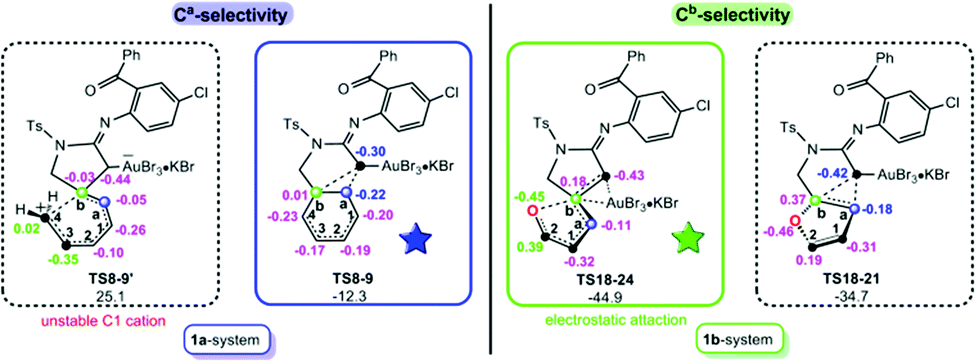 | ||
| Fig. 4 The schematic structures with NBO charges (in e) for competitive Ca and Cb selective-determining transition states in 1a- and 1b-systems. | ||
In summary, the mechanisms and origins of multiple nucleophilic site-selectivity of Au(III)-catalyzed annulations of substituted-anthranil 2M with N-benzyl ynamide 1a and N-furanylmethyl ynamide 1b, as well as that of unsubstituted-anthranil 2N and N-benzyl ynamide 1t, have been elucidated using DFT calculations. A unified and rational mechanism is presented in Fig. 2. After Au(III) coordination → 2M/2N N nucleophilic addition → N–O cleavage to afford gold carbene intermediate C, further Br(Au)-migration affords a common bromide intermediate D. Then, the three competitive reaction sites, Ca, Cb and Ct, selectively attack the Au-attached C atom, resulting in the distinct products. For 1a-/1b-annulations, a common key issue concerning a formal Cb-attack is established and involves an unprecedented “SN2-type N-donation/abstraction substitution” process. Subsequently, the reaction undertakes either Ca-migration → H-shift → Br-assisted H-shift → dehydration to form the Ca-attack product Pa in the 1a-system or C–O cleavage → Br-assisted H-shift to furnish the Cb-attack product Pb in the 1b-system. For Pt formation in the 1t system, the bromide intermediate, undergoes selective Ct-substitution with Br(Au), Ct-attached H-shift and dehydration.
For these annulation reactions, the present study indicates that the strength or weakness of the Br(Au) trans-influence dominates the nucleophilic selectivity for the Ca/Cb or Ct atoms. Furthermore, the Ca site-selectivity in the 1a-system can be attributed to the rather unstable terminal C cation in the ring-opening TS leading to Cb-attack, while the strong C⋯O electrostatic attraction in the crucial ring-expansion TS is responsible for the preferred Cb-selectivity in the 1b-system. In summary, we anticipate that the controlling factors involved in the nucleophilic site-selectivity observed for these reactions can be applied to other transition metal carbene-promoted reactions.
This work was jointly supported by the National Natural Science Foundation of China (21873055), the Natural Science Foundation of Shandong Province (ZR2019MB016 and ZR201709240033), Key Laboratory of Emergency and Trauma (Hainan Medical University), Ministry of Education (KLET-201903). TDJ wishes to thank the Royal Society for a Wolfson Research Merit Award and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University for support (2020ZD01).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS; (b) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS; (c) A. Furstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (d) L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953 CrossRef CAS; (e) L.-W. Ye, X.-Q. Zhu, R. L. Sahani, Y. Xu, P.-C. Qian and R.-S. Liu, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00348; (f) R. L. Sahani, L.-W. Ye and R.-S. Liu, Adv. Organomet. Chem., 2020, 73, 195 CrossRef; (g) E. Aguilar and J. Santamaría, Org. Chem. Front., 2019, 6, 1513 RSC.
- (a) S. Hahn, S. Koser, M. Hodecker, P. Seete, F. Rominger, O. Miljanic, A. Dreuw and U. H. F. Bunz, Chem. – Eur. J., 2018, 24, 6968 CrossRef CAS; (b) L. Ji, A. Friedrich, I. Krummenacher, A. Eichhorn, H. Braunschweig, M. Moos, S. Hahn, F. Geyer, O. Tverskoy, J. Han, C. Lambert, A. Dreuw, T. Marder and U. H. F. Bunz, J. Am. Chem. Soc., 2017, 139, 15968 CrossRef CAS; (c) Z. Zeng, H. Jin, X. Song, Q. Wang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem. Commun., 2017, 53, 4304 RSC.
- (a) M.-H. Tsai, C. Wang, A. S. K. Raj and R.-S. Liu, Chem. Commun., 2018, 54, 10866 RSC; (b) R. L. Sahani and R. Liu, Angew. Chem., Int. Ed., 2017, 56, 1026 CrossRef CAS; (c) R. L. Sahani and R.-S. Liu, Angew. Chem., Int. Ed., 2017, 56, 12736 CrossRef CAS; (d) G. Ung, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 758 CrossRef CAS.
- Z. Zeng, H. Jin, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 16549 CrossRef CAS.
- H. Jin, L. Huang, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 794 CrossRef CAS.
- (a) A.-H. Zhou, Q. He, C. Shu, Y.-F. Yu, S. Liu, T. Zhao, W. Zhang, X. Lu and L.-W. Ye, Chem. Sci., 2015, 6, 1265 RSC; (b) L. Li, T.-D. Tan, Y.-Q. Zhang, X. Liu and L.-W. Ye, Org. Biomol. Chem., 2017, 15, 8483 RSC; (c) H. Li, J. Liu, A. A. Ogunlana and X. Bao, Org. Chem. Front., 2017, 4, 1130 RSC; (d) S. S. Giri and R.-S. Liu, Chem. Sci., 2018, 9, 2991 RSC.
- A. A. Ogunlana and X. Bao, Chem. Commun., 2019, 55, 11127 RSC.
- (a) K. Wang, Y. Liu, Q. Wu, L. Liu, Y. Li, T. D. James, G. Chen and S. Bi, Mol. Catal., 2020, 480, 110647 CrossRef; (b) Y. Liu, X. Yang, L. Liu, H. Wang and S. Bi, Dalton Trans., 2015, 44, 5354 RSC; (c) Y. Liu, K. Wang, B. Ling, G. Chen, Y. Li, L. Liu and S. Bi, Catal. Sci. Technol., 2020, 10, 4219 RSC.
- (a) T. Li, W. Rong, T. Zhang and J. Li, ChemCatChem, 2020, 12, 5276 CrossRef CAS; (b) C. Zhu, L. Kou and X. Bao, Chin. J. Chem., 2020, 38, 57 CrossRef CAS; (c) A. K. Sahoo, R. Vanjari, S. Dutta, B. Prabagar and V. Gandon, Chem. – Asian J., 2019, 14, 4828 CrossRef.
- (a) M. J. Dewar, Angew. Chem., Int. Ed. Engl., 1971, 10, 761 CrossRef CAS; (b) M. Randić, Chem. Rev., 2003, 103, 3449 CrossRef; (c) Z. W. Davis-Gilbert, X. Wen, J. D. Goodpaster and I. A. Tonks, J. Am. Chem. Soc., 2018, 140, 7267 CrossRef CAS.
- It is confirmed by the IRC calculations that, in the 1t-system, the N–O bond breaks with simultaneous Br(Au)-migration, implying that the energy surface of the Br-migration is very flat. This fact can be indirectly corroborated by the corresponding step in the 1a- and 1b-systems, where the activation barriers are only 0.2 (Fig. 1) and 0.7 kcal mol−1 (Fig. S4, ESI†), respectively.
- In addition, to examine the contribution of the O⋯C2 electrostatic attraction to O–Cb cleavage, the furan O atom is replaced by a C atom (Fig. S6, ESI†). It was found that the ring-opening transition state is 2.8 kcal mol−1 higher in energy than the ring-expansion transition state, indicating that a strong electrostatic attraction between the O and C2 is a factor in the Cb-selectivity.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc07440c |
| This journal is © The Royal Society of Chemistry 2021 |